Gibson Assembly
Background and Design
Gibson Cloning is a technique of DNA construct assembly that allows one to join multiple linear segments into either one large linear segment or, if the segments contain the appropriate components and overlaps, an intact plasmid. This protocol follows the one-step isothermal assembly of overlapping dsDNA. In order to assemble segments of DNA via Gibson Cloning, they usually must contain at least 20bp of homology to the segment they are being joined to (Tm of overlapping region must be >= 48°C). Homology overlaps can vary in length from as few as 15bps up to 80bps -- efficacy depends on number of fragments assembled, as well as brand of "Gibson Mastermix" used. ie NEBuilder Hi-Fi DNA Assembly Mix will have a different optimal overlap length than NEB Gibson Master Mix and a different optimal length for homebrew Gibson Master Mix (recipe attached at bottom). Generally, 20bp overlap with proper a Tm is suitable. For example if one was to make a construct that was Seg1-Seg2-Seg3 from individual PCRs of Seg1, Seg2, and Seg3 (as indicated in the figure below) the 3' end of Seg1 would need at least 20bp of homology to the 5' end of Seg2 and Seg3 would require its 5' end to have at least 20bp of homology to Seg2. For constructing a plasmid, use a linearized vector backbone as one of your segments.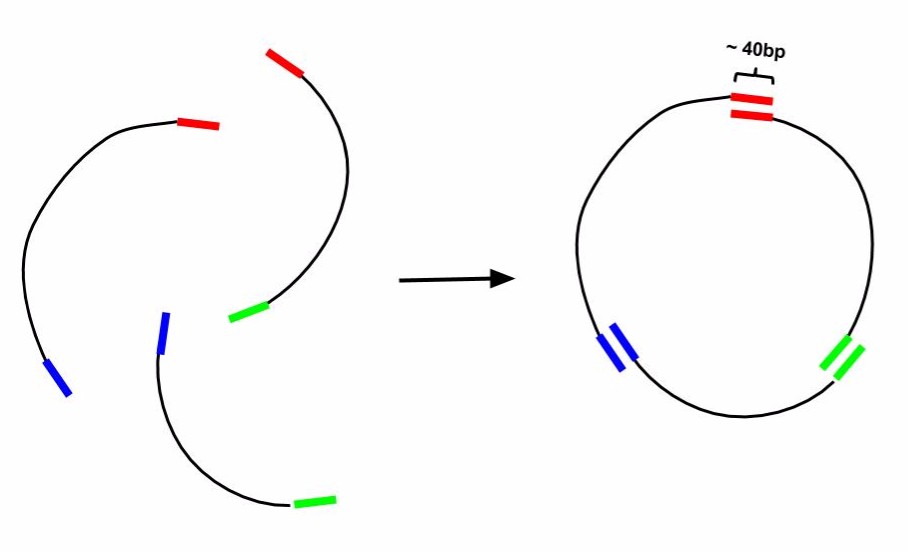
Supplies
If homemade master mix aliquots are available, and less than 1 year old:- 15µL aliquots or a 2X stock of Gibson master mix. Tubes are in both a 96 well holder, and a 50mL tube.
- Sterile, nuclease-free water
- Positive control: Positive Control DNA Mix (see below)
Primer Design Using Gibson
For a thorough discussion on the construction of primers for use in Gibson Assembly, please see the following publication: http://www.ncbi.nlm.nih.gov/pubmed/21601685.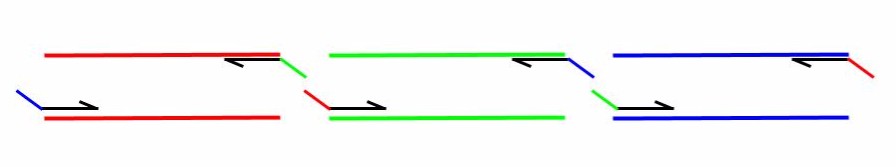 A note on primer design = Try to design your junctions to be in between the origin of replication or the selectable marker on the plasmid that you want to make. This will reduce your number of false-positives.
A note on primer design = Try to design your junctions to be in between the origin of replication or the selectable marker on the plasmid that you want to make. This will reduce your number of false-positives.
Protocol
- Use PCR to produce the DNA segments needed for assembling the new construct. Generally, it is best to use a high fidelity polymerase, such as Phusion, to amplify your Gibson fragments. See our following protocol for setting up a standard PCR reaction
- Confirm the success of each PCR by running 5µL of the reaction on an agarose gel. Gel purifying your fragments is always better than PCR purifying them - even if you only observe a single band on your gel.
- Mix 10ng-100ng of each of your DNA fragments together (such that their ratios are equimolar) into a 5µL total volume. Therefore, the length of each fragment, and the concentration of the fragments must be taken into account. Attached at the bottom of this page is an excel spread sheet calculator for an easy and accurate calculation of the amount of DNA needed.
- If using the 2X Gibson Master Mix from NEB, add 10µL of total DNA (containing all of your fragments) to 10µL of mix. If using the homemade Gibson mix (recipe at the bottom of this page), add 5µL of DNA to 15µL of mix. Be careful with pipetting small volumes.
- Mix well by pipetting.
- Incubate the reaction at 50°C for 1 hour.
- We generally use chemically competent cells for transforming Gibson reactions. For each Gibson reaction, you will do two transformations:
- Perform a 1:4 dilution of your Gibson reaction in nuclease-free water, add 4µL to 50µL of chemically competent cells
- Add 4µL of your Gibson reaction to 50µL competent cells without diluting
- Note = If electroporating, dilute the reaction 1:5 in water or you can PCR purify the reaction prior to transformation.
- Follow our transformation protocol for chemically competent cells
- Run an aliquot of your final reaction on a gel to verify the presence of your construct. It may be useful to make a 'no incubation' control to determine if the chemistry of the reaction happened.
Expected Results
The positive control should yield ampicillin resistant colonies when transformed into E. coli. You will see different efficiencies depending on the number of fragments in your reaction. Gibson reactions with a higher number of fragments tend to be less efficient, therefore you will likely see few colonies on your plates.Positive Control Construction and Usage
The Positive Control DNA Mix for Gibson Assembly consists of a two-piece assembly of pUC19. It is designed such that 5uL of the Positive Control DNA Mix is to be added to 15uL of Gibson Assembly Master Mix along side experimental reactions. Both pUC19 segments are between 1.3kb and 1.4kb in size. To construct the positive control reaction mix:- PCR amplify the two pUC19 fragments - fragment 1 (F1) and fragment 2 (F2).
- Use primers pUC19 F1 Gib FW (5'-CTCTTACTGTCATGCCATCCGTAAGATGCTTTTCTGTGACTGG-3') and pUC19 F1 Gib RV (5'-ACATGTTCTTTCCTGCGTTATCCCCTGATTCTGTGGATAA-3') to create F1 and pUC19 F2 Gib FW (5'-TTATCCACAGAATCAGGGGATAACGCAGGAAAGAACATGT-3') and pUC19 F2 Gib RV (5'-CCAGTCACAGAAAAGCATCTTACGGATGGCATGACAGTAAGAG-3').
- Use the following reaction mixture:
- 32.7µL dH2O
- 10µL 5X HF Buffer
- 1µL 10uM dNTP
- 2.5µL 10uM FW primer
- 2.5µL 10uM RV primer
- 1µL ~3ng/µL pUC19 template
- 0.3µL phusion polymerase
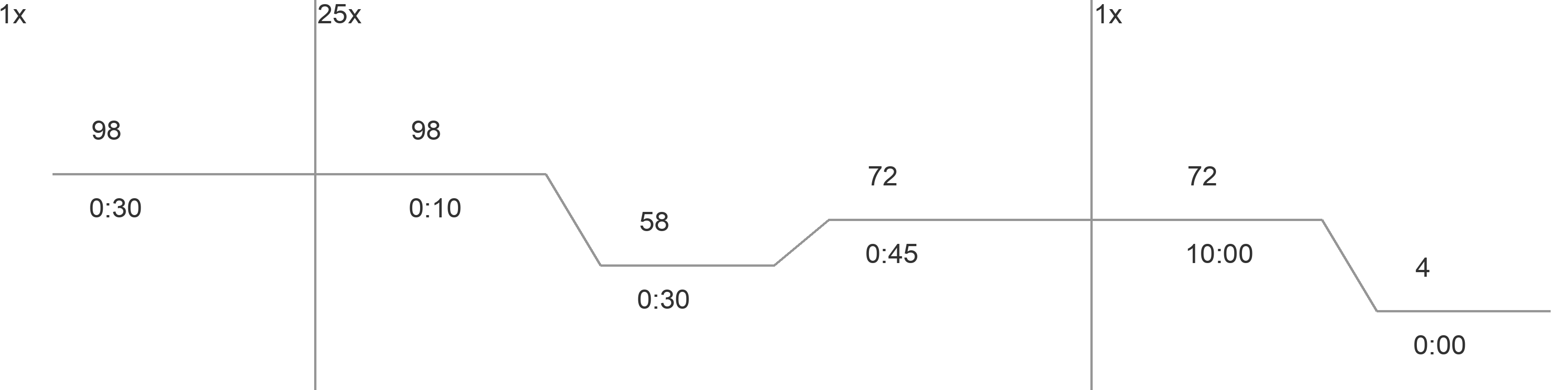
- Use the following PCR program for both F1 and F2 PCRs:
- 98°C - 0:30
- 98°C - 0:10
- 58°C - 0:30
- 72°C - 0:45
- Go to step 2, 25X
- 72°C - 10:00
- After the reaction completes, add 1µL of Dpn1 (20,000 U/mL) to each reaction, briefly vortex and spin down the reactions, and then incubate in the PCR machine at 37°C for 30 minutes. To test the effectiveness of your Dpn1, construct the above PCR reaction without Phusion polymerase, digest and purify it in parallel with the F1 and F2 PCRs, and transform it along side your other reactions. Any ampicillan-resistant transformants produced from from transforming this negative reaction indicate your Dpn1 digest was ineffective at digesting the pUC19 template. The solution to this is to use fresh Dpn1 in your digest, or gel extract F1 and F2 instead of using a Dpn1 digest and column purification to remove pUC19 template.
- Test the success of the PCR by gel electrophoresis and purify the reaction using a PCR cleanup kit, and elute with buffer or water without EDTA. Quantify the concentration of F1 and F2 purifications.
- Combine F1 and F2 PCR products to a final concentration of 2.8ng/µL of each fragment. Dilute with the elution buffer used in the PCR purification if needed. This final mixture is the Positive Control DNA Mix.
- Gibson_Master_Mix_Gibson_2011.xlsx: Gibson_rxn_master_mix *
- Bens_Gibson_Assembly.xlsx: gibson calc
- Gibson_Assembly_PCR.png:


| I | Attachment | History | Action | Size | Date | Who | Comment |
|---|---|---|---|---|---|---|---|
| |
Asset_543x.png | r1 | manage | 47.9 K | 2017-12-14 - 21:37 | UnknownUser | gibson method |
| |
Bens_Gibson_Assembly.xlsx | r1 | manage | 47.4 K | 2017-12-14 - 21:50 | UnknownUser | gibson calc |
| |
GibsonPrimerDesign.jpg | r1 | manage | 14.8 K | 2016-12-16 - 15:33 | UnknownUser | |
| |
Gibson_Assembly_PCR.png | r1 | manage | 37.5 K | 2021-10-22 - 16:02 | CameronRoots | |
| |
Gibson_Master_Mix_Gibson_2011.xlsx | r1 | manage | 10.9 K | 2017-09-05 - 21:32 | UnknownUser | Gibson_rxn_master_mix |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Barrick Lab > ProtocolList > ProtocolsGibsonCloning