Gel Electrophoresis
Overview
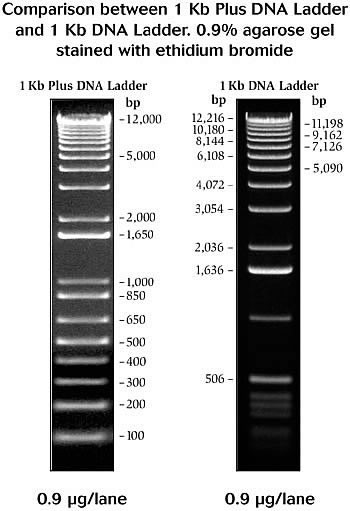
Gel electrophoresis separates DNA fragments based on size. Electric current moves the negatively charged DNA through the gel, which slows down larger pieces of DNA and allows smaller pieces to move faster. Bands on the gel can be compared to a "ladder" of DNA fragments of known lengths (Fig. 1) run in a separate well to determine the size of each piece of DNA.Fig. 1 NEB 1 kb and 1 kb+ ladders.
Making the Gel
Most PCRs can be analyzed on a standard 1% agarose gel (1 g of agarose per 100 mL of buffer). Changing the concentration can be helpful for separating very large (use a 0.75% agarose gel) or very small (use a 2% agarose gel) DNA molecules. For a standard 1% 50 ml gel: 1. Add 0.5 g agarose and 50 mL TAE buffer to a 125 mL flask and heat in the microwave (on high power) for 1:30 minutes. While the microwave is running you can set up the gel mold and well combs. 2. After heating add 2.5 μL SYBR Safe and swirl to mix. 3. Pour the liquid from the flask into the mold and let it solidify (approximately 20-30 minutes). 4. Once the gel has solidified, remove the comb carefully so as not to break the walls of the wells. Then:- If using immediately place the gel into a rig. Proceed to "Loading and Running the Gel," below.
- If not being used immediately, the gel can be saved for a few days by wrapping it tightly in cellophane and storing in the 4 °C fridge.
Loading and Running the Gel
DNA samples should be mixed with a loading dye before analyzing to help load them into the wells. Each sample and the ladder will then be pipetted into a separate well in the gel. Steps: 1. Pour TAE buffer into the rig until the gel is thoroughly submerged. 2. Load 6 μL of 1 kb+ ladder working solution into the first well (dye is already combined with our ladder*). 3. Cut out a piece of parafilm to mix the loading dye and DNA samples. For each sample pipette a 1 μL drop of loading dye onto the parafilm. Then, mix 5 μL of PCR product into a drop of dye and pipette up and down to mix. Finally, pipette the dye/sample mix into an empty well. Record which lane each sample is loaded into. 4. Once all samples are loaded, attach the lid to the rig with the negative (black) electrode at the top and the positive (red) electrode at the bottom. (NOTE: always make sure that the current is off or paused before inserting or removing cords from the power supply or rig). 5. Set the voltage to 120 V and run the gel for 20-30 minutes (you can set a timer on the machine itself by clicking on the clock logo. The run will stop when the time runs out). It is advisable to check up on the gel from time to time to make sure that it is proceeding normally. The tracking dye should migrate towards the bottom of the gel. 6. When the gel seems to be completed, pause/stop the voltage, disconnect and remove the lid, and take the gel (in its tray) to the Bio-Rad gel imaging machine. Note: Close PCR tubes when not being used. PCR products tend to dry up when exposed to air, leaving a lower volume that has a higher concentration of DNA. *Our ladder working stock in the 4 °C fridge is colored purple from the loading dye and has been diluted as described here. Do not use the colorless undiluted stock, the bands will be too dense.Imaging and Analyzing the Gel
Steps: 1. Remove the gel from the tray and place it in the imager. Open the Image Lab program. 2. Select "SYBR Safe" from the "Nucleic Acids Gels" drop-down menu (unless using a different fluorescent dye), uncheck the "Highlight saturated pixels" box, then click "Position Gel." Center your gel in the viewer and zoom in to optimize your view of the gel. 3. Once centered, close the imager door and click "Run Protocol." 4. Modify the image as you see fit**. 5. Save the image in Documents/[your name]. Note that the program saves files as .scn, which can only be opened by Image Lab and related programs. For a simpler version which can be viewed from any computer, select snapshot, then save a common file type (ex: .jpg). If you want a physical copy you can also print the image. **Image Lab contains several tools to aid in analyzing the gel image. Hitting the change contrast button allows manipulation of the brightness of the image, which may be helpful to clearly see faint bands. By clicking Lanes and Bands, then Automatic, the program puts labeled lanes where it perceives them. Bands can similarly be applied to the image.Post-staining Gels
Adding DNA dyes to agarose gels after electrophoresis (post-staining) can be advantageous when very crisp bands are needed (publication, low amount of DNA, etc). Adding nucleic acid dye to the molten agarose can slightly inhibit DNA migration and cause bands to bend. Post-staining can reduce bending, increase DNA signal, and reduce background signal. Steps: 1. After gel electrophoresis, place gel into small tray. 2. Add 15 µL of dye into 50 mL buffer (typically x1 TAE) and add to tray. 3. Gently agitate for 30 minutes. 4. Remove buffer+dye. 5. Wash gel with ddH2O 2-3 times. 6. Image gel. *Notes:* 1. 15 µL of GelRed into 50 mL buffer is the correct dilution. 2. SYBR-safe gel documentation notes that 50 µL of dye should be added to 50 mL buffer, though 15 µL of dye into 50 mL is likely sufficient (not tested). 3. Typically, dye is diluted in the same buffer that the gel is made of/was run in (e.g.: x1 TAE). 4. Any volume of buffer+dye can be used, though it must completely cover the gel. 5. Post-staining buffer can be reused up to 5 times by storing in dark storage container (e.g.: a 50 mL conical tube covered with aluminum foil). Barrick Lab > ProtocolList > ProceduresStandardAgaroseGel