16S rRNA Sequencing to Identify Unknown Microbes
Ever wonder what that contaminant in your culture is? Need to accurately identify an environmental isolate?Overview
- Direct PCR of culture or isolation of DNA from culture before PCR.
- PCR using universal primers.
- Agarose gel to confirm PCR worked correctly.
- Excise correct bands from gel and purify DNA.
- Prepare and submit samples for sequencing.
PCR Reaction
For PCR we use universal primers U341F and UA1406R that amplify an approximately 1 kb stretch of rDNA and should work for nearly any bacterial or archaeal sequence [1].| primer | sequence |
|---|---|
| U341F | CCTACGGGRSGCAGCAG |
| UA1406R | ACGGGCGGTGWGTRCAA |
It is generally preferred to isolate genomic DNA from your culture before doing the 16s RNA PCR (using the Invitrogen PureLink kit, for example). This results in a cleaner PCR product and generally a better sequencing result.
After DNA is isolated, use a Qubit to find the concentration of DNA. The desired amount of gDNA template for PCR is 0.1 ng/ul (final template concentration) so for a 30 ul PCR reaction use 3 ng of DNA.
The PCR cycle is as follows:
| 94 | 10 min |
| 94 | 30 sec |
| 54 | 30 sec |
| 72 | 1 min |
After PCR, the products need to be run on an agarose gel to confirm correct product (around 1kb). A 1.5% or 2% gel works the best for this. If the bands are clean, then you can do a PCR cleanup spin column and submit the DNA for two different sequencing reactions (one with each of your PCR primers). If the bands are not clean, you may be able to excise the band and The bands should be clean to excise the correct bands from the gel.
After cutting the bands out of gel, clean using Zymo gel extraction kit or equivalent.
Proceed to send for Sanger Sequencing.
Analysis
Use one of these tools to assign your species:- Targeted Loci BLAST - select the appropriate database for your samples. Usually: rRNA/ITS databases > 16S ribosomal RNA sequences (Bacteria and Archaea)
- BLAST result for Pseudomonas syringae:
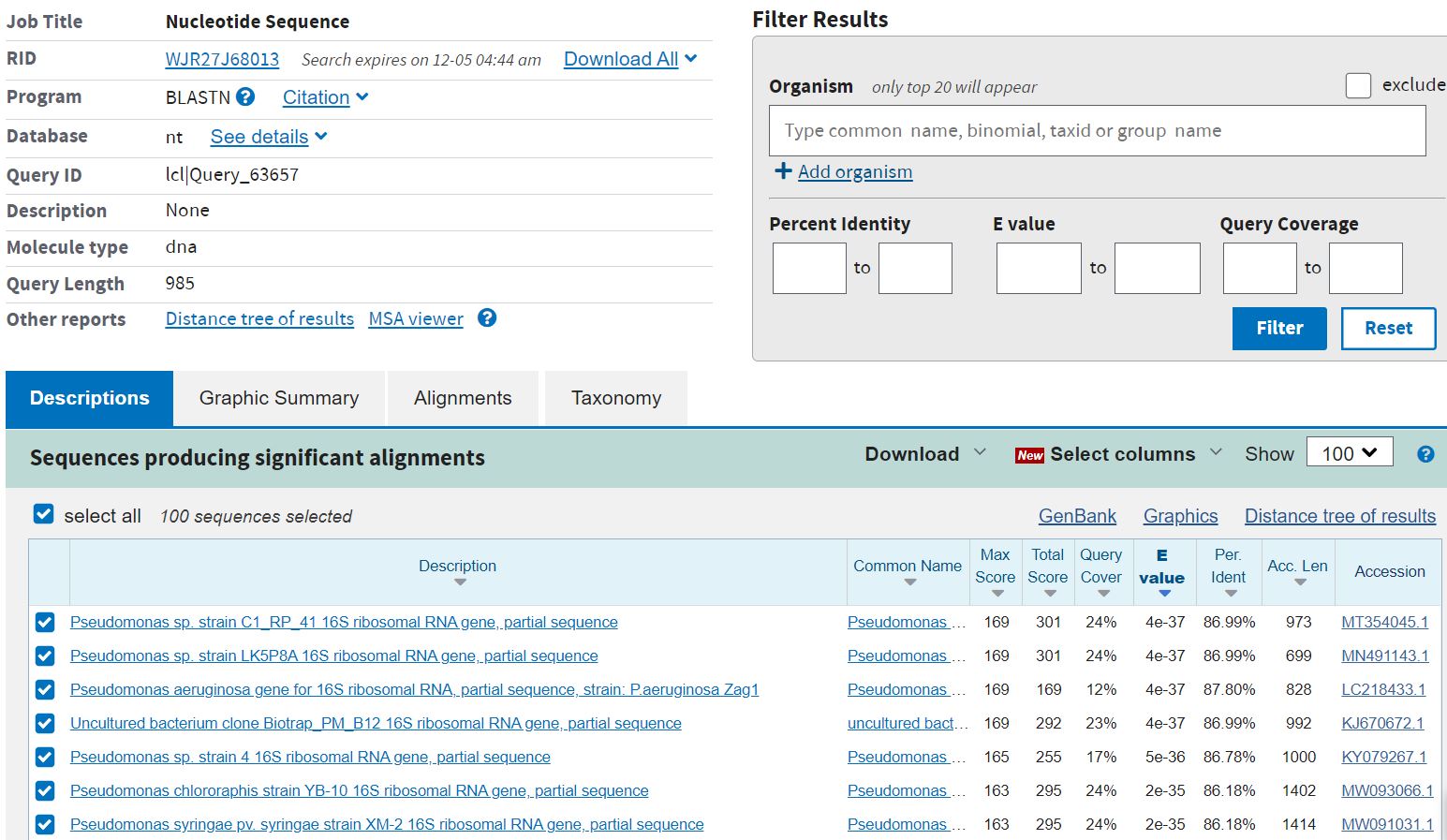
- BLAST result for Pseudomonas syringae:
References
1. Baker, G.C., Smith, J.J. & Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55, 541-55 (2003).

| I | Attachment | History | Action | Size | Date | Who | Comment |
|---|---|---|---|---|---|---|---|
| |
BLAST.JPG | r1 | manage | 217.2 K | 2020-12-03 - 21:15 | UnknownUser | BLAST result for iPseudomonas syringae/i |
| |
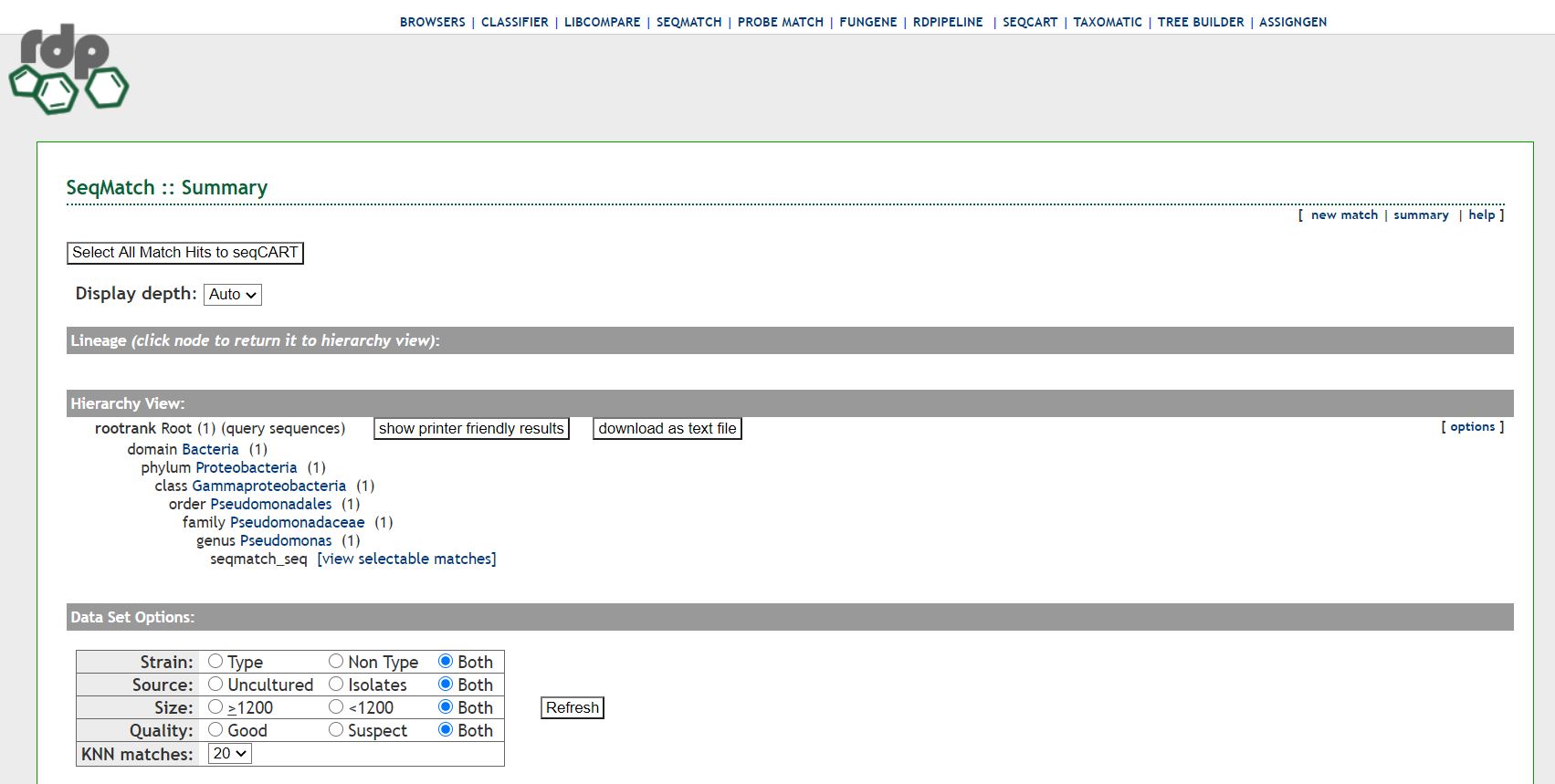
Ribosomal_Database_Project.JPG | r1 | manage | 135.9 K | 2020-12-03 - 21:10 | UnknownUser | SeqMatch results for Pseudomonas syringae |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Barrick Lab > ProtocolList > Protocols16SSequencing