Difference: ProtocolsFluorescentProteinEvolutionaryStability (1 vs. 11)
Revision 112018-03-22 - JeffreyBarrick
Evolutionary Stability of Fluorescent Protein or Chromoprotein ExpressionProcedure for Studying the Evolutionary Stability of a Chromoprotein in TOP10 and/or MDS42 CellsThis procedure can also be used for studying any similar genetic device, such as a device encoding a fluorescent protein. The specifics below are from an experiment conducted by Riya Sreenivasan, an FRI/iGEM student during summer 2016.
The above image shows the blue chromoprotein construct (described below) over the course of 6 days, using the procedure below. Day 0Streak out frozen stocks of two strains on two separate plates containing LB and the appropriate antibiotic.
Day 1Today is Day 1 of growing your strain(s) in liquid culture. These first culture tubes are dubbed "Day 1" culture tubes.
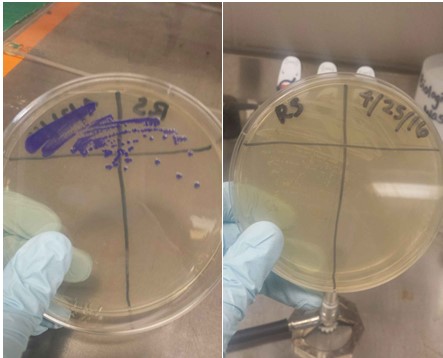 Plates of two different Blue Chromoprotein (BCP) strains streaked out. The one is still expressing high levels of BCP. The other strain is not. Side note: BCP is commonly referred to as "AmilCP".
Day 1 Procedure: Starting your Overnight (O/N) cultures from colonies.
Plates of two different Blue Chromoprotein (BCP) strains streaked out. The one is still expressing high levels of BCP. The other strain is not. Side note: BCP is commonly referred to as "AmilCP".
Day 1 Procedure: Starting your Overnight (O/N) cultures from colonies.
Day 2Your cultures will need to grow in the incubator overnight. We suggest for at least 20 hours. Record this length of time and attempt to repeat this exact length of time as closely as possible. It is okay if you over shoot by an hour or two, but make sure the overnight culture is fully saturated. NOTE: Saturation of these cultures should happen in less than 20 hrs, but better safe than sorry.
Day 3 → until completionRepeat the procedure for Day 2, preparing new culture tubes with labeled with the respective days. Record the absorbance or fluorescence readings from the previous day as you did on Day 2. This data should be recorded online and in your notebooks. UT Box is our preferred storage area. Record how many generations passed between each day. After 7-10+ days, or after a time when the phenotype of the cultures visibly diminishes, you should stop carrying forward the overnight cultures. *FRI STUDENTS: You will want to check this with Dr. Mishler or a mentor prior to stopping. In general, we will want to see that the color has declined and stayed declined for at least 2 consecutive days. [[STOPPED HERE BY DR. MISHLER ON 12/15/2016. Will continue later from Riya's 2016 protocol. - Dr. M.]]] Day completion = Day X In consultation with Dr. Mishler, stop growing cultures once the color has significantly diminished.Procedure: Growth
Procedure: Measurement Measurement by Plating
There are essentially two ways to do this by plating, either by spreading an inoculum across the whole plate or by spotting inocula as serial dilutions. Both involve counting colonies and the readout is the ratio of fluorescent colonies divided by the number of total colonies. Though spreading consumes a significantly higher number of plates, it also allows detection of smaller changes in fluorescence, as typically 30-300 colonies can be counted on such plates while only about 6-60 colonies can be counted during spotting.
| ||||||||
| Deleted: | ||||||||
| < < | Measurement by Next-Gen SequencingExpected Results | |||||||
Authors and Acknowledgements
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
Revision 102016-12-15 - DennisMishler
| ||||||||
| Changed: | ||||||||
| < < | Evolutionary Stability of Fluorescent Protein Expression | |||||||
| > > | Evolutionary Stability of Fluorescent Protein or Chromoprotein Expression | |||||||
|
| ||||||||
| Changed: | ||||||||
| < < | This procedure is to monitor the decay of a genetic device that outputs a visible signal, such as GFP fluorescence. As a microbe replicates, it frequently accumulates mutations in the genetic devices that we have introduced. The mutations that inactivate gene expression, such as loss of a start codon or loss of a promoter can lead to a lower burden (or metabolic load) on the cell. These cells containing inactivated genetic devices will commonly out compete the cells that contain functional genetic devices. If said genetic device yielded a fluorescent signal, then this will lead to a loss in fluorescent signal over time as the cells with the inactivated genetic device sweep the population. | |||||||
| > > | This procedure is to monitor the decay of a genetic device that outputs a visible signal, such as GFP fluorescence. As a microbe replicates, it frequently accumulates mutations in the genetic devices that we have introduced. The mutations that inactivate gene expression, such as loss of a start codon or loss of a promoter can lead to a lower burden (or metabolic load) on the cell. These cells containing inactivated genetic devices will commonly out compete the cells that contain functional genetic devices. If said genetic device yielded a fluorescent signal, then this will lead to a loss in fluorescent signal over time as the cells with the inactivated genetic device sweep the population. There are many different types of mutations. Some can completely knock out protein expression, others may only partially reduce expression or signal. Thus, studying individual cells within as well as the entire cell culture population may be necessary. | |||||||
Procedure for Studying the Evolutionary Stability of a Chromoprotein in TOP10 and/or MDS42 CellsThis procedure can also be used for studying any similar genetic device, such as a device encoding a fluorescent protein. The specifics below are from an experiment conducted by Riya Sreenivasan, an FRI/iGEM student during summer 2016. | ||||||||
| Changed: | ||||||||
| < < | ||||||||
| > > | ||||||||
| Added: | ||||||||
| > > |
The above image shows the blue chromoprotein construct (described below) over the course of 6 days, using the procedure below. | |||||||
Day 0Streak out frozen stocks of two strains on two separate plates containing LB and the appropriate antibiotic.
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
| Added: | ||||||||
| > > | Day 0 Procedure: | |||||||
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
Day 1Today is Day 1 of growing your strain(s) in liquid culture. These first culture tubes are dubbed "Day 1" culture tubes.
| ||||||||
| Added: | ||||||||
| > > |
Plates of two different Blue Chromoprotein (BCP) strains streaked out. The one is still expressing high levels of BCP. The other strain is not. Side note: BCP is commonly referred to as "AmilCP".
Day 1 Procedure: Starting your Overnight (O/N) cultures from colonies. | |||||||
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
Day 2Your cultures will need to grow in the incubator overnight. We suggest for at least 20 hours. Record this length of time and attempt to repeat this exact length of time as closely as possible. It is okay if you over shoot by an hour or two, but make sure the overnight culture is fully saturated. NOTE: Saturation of these cultures should happen in less than 20 hrs, but better safe than sorry. | ||||||||
| Deleted: | ||||||||
| < < | [[STOPPED HERE BY DR. MISHLER ON 8/25/2016. Will continue later from Riya's 2016 protocol. - Dr. M.]]] | |||||||
| Added: | ||||||||
| > > | ||||||||
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
| Added: | ||||||||
| > > |
| |||||||
| ||||||||
| Added: | ||||||||
| > > |
| |||||||
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
| Added: | ||||||||
| > > |
| |||||||
| Added: | ||||||||
| > > | Day 3 → until completionRepeat the procedure for Day 2, preparing new culture tubes with labeled with the respective days. Record the absorbance or fluorescence readings from the previous day as you did on Day 2. This data should be recorded online and in your notebooks. UT Box is our preferred storage area. Record how many generations passed between each day. | |||||||
| Added: | ||||||||
| > > | After 7-10+ days, or after a time when the phenotype of the cultures visibly diminishes, you should stop carrying forward the overnight cultures. *FRI STUDENTS: You will want to check this with Dr. Mishler or a mentor prior to stopping. In general, we will want to see that the color has declined and stayed declined for at least 2 consecutive days. | |||||||
| Added: | ||||||||
| > > | [[STOPPED HERE BY DR. MISHLER ON 12/15/2016. Will continue later from Riya's 2016 protocol. - Dr. M.]]] Day completion = Day X In consultation with Dr. Mishler, stop growing cultures once the color has significantly diminished. | |||||||
| Added: | ||||||||
| > > | ||||||||
Procedure: Growth
Procedure: Measurement Measurement by Plating
There are essentially two ways to do this by plating, either by spreading an inoculum across the whole plate or by spotting inocula as serial dilutions. Both involve counting colonies and the readout is the ratio of fluorescent colonies divided by the number of total colonies. Though spreading consumes a significantly higher number of plates, it also allows detection of smaller changes in fluorescence, as typically 30-300 colonies can be counted on such plates while only about 6-60 colonies can be counted during spotting.
| ||||||||
| Added: | ||||||||
| > > | Authors and Acknowledgements
| |||||||
| ||||||||
| Added: | ||||||||
| > > |
| |||||||
Revision 92016-09-02 - JeffreyBarrick
Evolutionary Stability of Fluorescent Protein ExpressionProcedure for Studying the Evolutionary Stability of a Chromoprotein in TOP10 and/or MDS42 CellsThis procedure can also be used for studying any similar genetic device, such as a device encoding a fluorescent protein. The specifics below are from an experiment conducted by Riya Sreenivasan, an FRI/iGEM student during summer 2016.Day 0Streak out frozen stocks of two strains on two separate plates containing LB and the appropriate antibiotic.
Day 1Today is Day 1 of growing your strain(s) in liquid culture. These first culture tubes are dubbed "Day 1" culture tubes.
Day 2Your cultures will need to grow in the incubator overnight. We suggest for at least 20 hours. Record this length of time and attempt to repeat this exact length of time as closely as possible. It is okay if you over shoot by an hour or two, but make sure the overnight culture is fully saturated. NOTE: Saturation of these cultures should happen in less than 20 hrs, but better safe than sorry. [[STOPPED HERE BY DR. MISHLER ON 8/25/2016. Will continue later from Riya's 2016 protocol. - Dr. M.]]]
Procedure: Growth
Procedure: Measurement Measurement by Plating
There are essentially two ways to do this by plating, either by spreading an inoculum across the whole plate or by spotting inocula as serial dilutions. Both involve counting colonies and the readout is the ratio of fluorescent colonies divided by the number of total colonies. Though spreading consumes a significantly higher number of plates, it also allows detection of smaller changes in fluorescence, as typically 30-300 colonies can be counted on such plates while only about 6-60 colonies can be counted during spotting.
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
Revision 82016-08-25 - DennisMishler
Evolutionary Stability of Fluorescent Protein Expression | ||||||||
| Changed: | ||||||||
| < < | This procedure is to monitor the decay of a genetic device that outputs GFP fluorescence. As a microbe replicates, it accumulates mutations that lead to a loss in fluorescent signal, and these mutant cells usually outcompete cells with fully functioning copies of the device. | |||||||
| > > | This procedure is to monitor the decay of a genetic device that outputs a visible signal, such as GFP fluorescence. As a microbe replicates, it frequently accumulates mutations in the genetic devices that we have introduced. The mutations that inactivate gene expression, such as loss of a start codon or loss of a promoter can lead to a lower burden (or metabolic load) on the cell. These cells containing inactivated genetic devices will commonly out compete the cells that contain functional genetic devices. If said genetic device yielded a fluorescent signal, then this will lead to a loss in fluorescent signal over time as the cells with the inactivated genetic device sweep the population. | |||||||
| Added: | ||||||||
| > > |
Procedure for Studying the Evolutionary Stability of a Chromoprotein in TOP10 and/or MDS42 CellsThis procedure can also be used for studying any similar genetic device, such as a device encoding a fluorescent protein. The specifics below are from an experiment conducted by Riya Sreenivasan, an FRI/iGEM student during summer 2016.Day 0Streak out frozen stocks of two strains on two separate plates containing LB and the appropriate antibiotic.
Day 1Today is Day 1 of growing your strain(s) in liquid culture. These first culture tubes are dubbed "Day 1" culture tubes.
Day 2Your cultures will need to grow in the incubator overnight. We suggest for at least 20 hours. Record this length of time and attempt to repeat this exact length of time as closely as possible. It is okay if you over shoot by an hour or two, but make sure the overnight culture is fully saturated. NOTE: Saturation of these cultures should happen in less than 20 hrs, but better safe than sorry. [[STOPPED HERE BY DR. MISHLER ON 8/25/2016. Will continue later from Riya's 2016 protocol. - Dr. M.]]]
| |||||||
Procedure: Growth
Procedure: Measurement Measurement by Plating
There are essentially two ways to do this by plating, either by spreading an inoculum across the whole plate or by spotting inocula as serial dilutions. Both involve counting colonies and the readout is the ratio of fluorescent colonies divided by the number of total colonies. Though spreading consumes a significantly higher number of plates, it also allows detection of smaller changes in fluorescence, as typically 30-300 colonies can be counted on such plates while only about 6-60 colonies can be counted during spotting.
| ||||||||
| META FILEATTACHMENT | attachment="fluorescence_for_wiki-1.png" attr="" comment="An example scatterplot generated to represent loss in fluorescence measured by plating." date="1458915164" name="fluorescence_for_wiki-1.png" path="fluorescence for wiki-1.png" size="166699" stream="fluorescence for wiki-1.png" tmpFilename="/usr/tmp/CGItemp44896" user="LucyLeblanc" version="1" |
|---|
Revision 72016-04-07 - DaciaLeon
Evolutionary Stability of Fluorescent Protein ExpressionProcedure: Growth
Procedure: Measurement Measurement by Plating
There are essentially two ways to do this by plating, either by spreading an inoculum across the whole plate or by spotting inocula as serial dilutions. Both involve counting colonies and the readout is the ratio of fluorescent colonies divided by the number of total colonies. Though spreading consumes a significantly higher number of plates, it also allows detection of smaller changes in fluorescence, as typically 30-300 colonies can be counted on such plates while only about 6-60 colonies can be counted during spotting.
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
Measurement by Next-Gen SequencingExpected Results
| ||||||||
Revision 62016-03-25 - LucyLeblanc
Evolutionary Stability of Fluorescent Protein ExpressionProcedure: Growth
Procedure: Measurement Measurement by Plating
There are essentially two ways to do this by plating, either by spreading an inoculum across the whole plate or by spotting inocula as serial dilutions. Both involve counting colonies and the readout is the ratio of fluorescent colonies divided by the number of total colonies. Though spreading consumes a significantly higher number of plates, it also allows detection of smaller changes in fluorescence, as typically 30-300 colonies can be counted on such plates while only about 6-60 colonies can be counted during spotting.
| ||||||||
| Added: | ||||||||
| > > | Expected ResultsBoth spotting and spreading yield the same output in terms of data, that is, fluorescence is presented as a proportion of fluorescent colonies over total colonies as a function of time. This data should thus be presented as a scatterplot. Furthermore, fluorescence by these measurements is highly stable; after three weeks, over 90% of the colonies seem to remain fluorescent (at least in preliminary tests). Here is example output graphed via ggplot2 in R: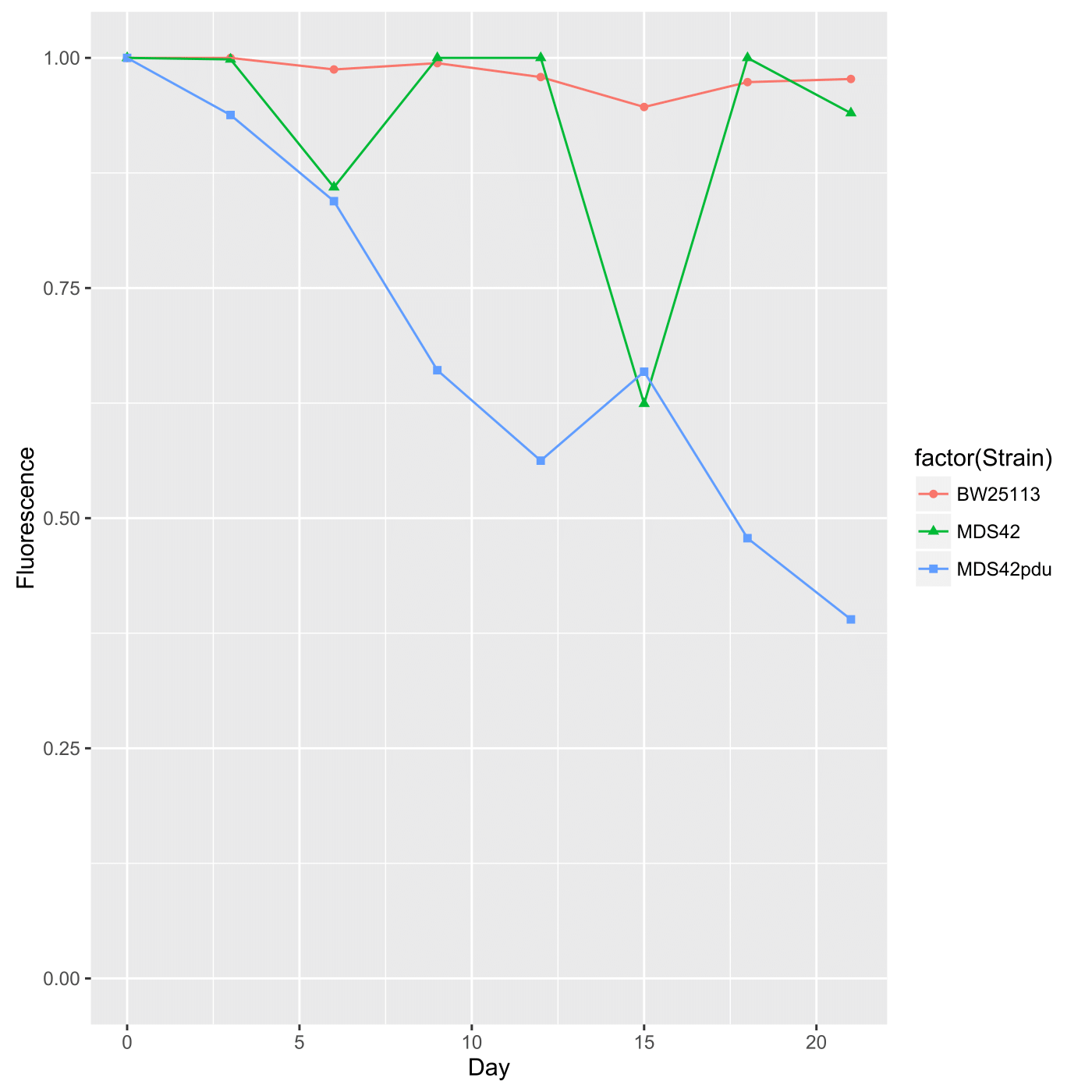 This was generated via the following code in the terminal using R (v. 3.2.3), which has been commented so that you may use it as a backbone if you are unfamiliar with R:
install.packages("ggplot2")
This was generated via the following code in the terminal using R (v. 3.2.3), which has been commented so that you may use it as a backbone if you are unfamiliar with R:
install.packages("ggplot2")#If you are running ggplot2 for the first time, take note that it is not included automatically when you install R; thus it must be installed manually. library("ggplot2") #ggplot2 is not loaded by default when you start up R; this is the standard way of loading it. data <- read.csv("yourfilename.csv", header = TRUE, sep = ",") #This reads your input file and deposits it into 'data', given that the data points are separated by commas and that there is a header of column names. ggplot(data, aes(x = Day, y = Fluorescence, color = factor(Strain), shape = factor(Strain))) + geom_point() + geom_line() + ylim(0, 1.0) #This invokes ggplot2 and graphs fluorescence (y) as a function of time (x). #Points on the graph can color-coded (color = factor(Strain)) and shape-coded (shape = factor(Strain)) for clarity if your data points originate from different samples. Since fluorescence here is represented by a proportion such that 0 ≤ y ≤ 1, ylim(0, 1.0) sets the boundaries of the y-axis. For more guidance on R, helpful resources can be found for aesthetics and formatting of axes. | |||||||
Troubleshooting:
Measurement by Microplate Reader
Troubleshooting:
Measurement by Flow CytometryNote
Measurement by Next-Gen SequencingExpected Results | ||||||||
| Added: | ||||||||
| > > |
| |||||||
Revision 52016-03-24 - JeffreyBarrick
Evolutionary Stability of Fluorescent Protein Expression | ||||||||
| Changed: | ||||||||
| < < | This procedure is to monitor the decay of a genetic device that outputs GFP fluorescence as a microbe replicates, it accumulates mutations that lead to a loss in fluorescent signal, and these mutant cells outcompete cells with fully functioning copies of the device. | |||||||
| > > | This procedure is to monitor the decay of a genetic device that outputs GFP fluorescence. As a microbe replicates, it accumulates mutations that lead to a loss in fluorescent signal, and these mutant cells usually outcompete cells with fully functioning copies of the device. | |||||||
Procedure: Growth
Procedure: Measurement Measurement by Plating | ||||||||
| Changed: | ||||||||
| < < | There are essentially two ways to do this by plating, either by spreading an inoculum across the whole plate or by spotting inocula as serial dilutions. Both involve counting colonies and the readout is the ratio of fluorescent colonies divided by the number of total colonies. Though spreading consumes a significantly higher number of plates, it also allows detection of smaller changes in fluorescence, as typically 30-300 colonies can be counted on such plates while only about 6-60 colonies can be counted during spotting. | |||||||
| > > | There are essentially two ways to do this by plating, either by spreading an inoculum across the whole plate or by spotting inocula as serial dilutions. Both involve counting colonies and the readout is the ratio of fluorescent colonies divided by the number of total colonies. Though spreading consumes a significantly higher number of plates, it also allows detection of smaller changes in fluorescence, as typically 30-300 colonies can be counted on such plates while only about 6-60 colonies can be counted during spotting. | |||||||
| Changed: | ||||||||
| < < | Spreading 1) Estimate the cfu/ml of the overnight culture via OD600 measurement. There is a web calculator available here. Note that to measure OD600 accurately you may have to dilute your culture as this is most accurate in the range of OD600 in the range of 0.1 to 1.0. 2) After estimation, dilute the culture in saline by taking 100 μL, adding it to 10 mL of saline, vortexing, and repeating the dilution process as needed. The ultimate goal is to dilute the cfu/ml to a point where spreading 50-100 μL would yield about 30-300 colonies. 3) Add sterile glass beads to LB agar plates with antibiotic, then place the appropriate amount of the diluted culture on the beads. 4) Shake the plates back and forth to ensure that the inoculum is spread uniformly across the surface of the agar. 5) Incubate at 37°C for 24 h. 6) Count the total number of colonies overall. If desired, you may use a colony-counting software such as OpenCFU, but it may be easier to simply do it by hand. If you do decide to use such software, please double-check the automated counts to eliminate any false positives and false negatives. 7) Count the total number of nonfluorescent colonies using blue light. Remember to place the orange filter on top and, if desired, wear the orange safety glasses. 8) Now, to calculate the ratio, subtract the number of nonfluorescent colonies from the number of total colonies. This will yield the number of fluorescent colonies. Then divide by the total number of colonies. 9) Plot the ratio as a function of time over several days. The expected result is a significant drop in this ratio over the course of a few days representing the decay of the genetic device. | |||||||
| > > | Spreading
| |||||||
| Changed: | ||||||||
| < < | Spotting Note: This protocol was adapted from Thomas, Sekhar, Upreti, Mujawar, & Pasha (2015). A convenient flowchart of the protocol (though altered) can be found here. 1) It is no longer necessary to estimate the cfu/ml of the overnight culture, as each plate is capable of enumerating colonies over a range of 6 orders of magnitude as opposed to the above method. Instead of diluting the overnight culture in 10 ml of saline, set up a serial dilution series of sterile microfuge tubes each containing 900 μl of saline. Each tube represents a tenfold dilution (and thus should be labeled -1, -2, -3, etc.) and I recommend diluting out to 10^-7. Contrary to what the paper advises, I do not recommend diluting the overnight culture to an OD600 of 0.1; it is unnecessary. 2) Place 100 μl of the overnight culture into the microfuge tube representing 10^-1 and vortex. Please note that taking 100 μl from this would actually be a 10^-2 dilution (the overnight culture is first diluted tenfold after being added to the saline, then you only take 1/10th of the dilution to plate). This will be accounted for in the final formula to back calculate cfu/ml (if desired). 3) Label your LB agar plates with antibiotic by dividing most of the plate into 6 sections. Each section of the plate will be used to spot a different dilution of the same culture. Thus, the six sections may be labeled -2, -3, -4, -5, -6, and -7, for example. 4) Spot 10 μl of each dilution in triplicate onto the corresponding section of the plate. The paper recommends 20 ul but that seems to cause the spots to run into one another and it also takes longer to dry, increasing the chance for contamination. 5) Dry out the plate in the 30°C or 37°C incubator with the lid slightly off to allow moisture to evaporate. This part can take 15-20 mins depending on temperature and how moist the agar itself is. 6) Place the lid back on fully and incubate at 37°C for 24 h. The resulting plate should look like the example plate on the left here. 7) Calculate the ratio of fluorescent to nonfluorescent colonies. 8) Plot the ratio as a function of time over several days. | |||||||
| > > | SpottingNote: This protocol was adapted from Thomas, Sekhar, Upreti, Mujawar, & Pasha (2015).A convenient flowchart of the protocol (though altered) can be found here.
| |||||||
| Changed: | ||||||||
| < < | Troubleshooting: a) The incubation period is around 24 h because colonies that are too small may not appear fluorescent (even if the genetic device is intact) and thus act as false negatives. However, growth rates can vary and you should check on colony growth before 24 h if colonies have grown too large and start to overlap. b) Fluorescent colonies can stay fluorescent for a couple of weeks in the fridge, so feel free to store them at 4°C and count them later if desired. c) If there is large variation between technical replicates, ensure you are vortexing for a few seconds after every dilution step. d) While drying the inocula during the spotting method, even though the plate is somewhat open and exposed for a period of time, it is unlikely that you will get any contamination. If this turns out to be a problem, you may also dry your plates in a biosafety cabinet which will take longer due to lower temperature but reduce the risk of contamination. | |||||||
| > > | Troubleshooting:
| |||||||
Measurement by Microplate Reader | ||||||||
| Changed: | ||||||||
| < < | 1) Plan out which wells will contain what sample. You can print out one of the 96 well plate templates available online. Generally, each condition should be done in quadruplicate (or triplicate depending on space concerns). 2) After vortexing your sample, aliquot 100μL at a time into each well. You should use the appropriate type of plate for your application: black walled, clear bottom for fluorescence (white walled, clear bottom is generally for luminescence and completely clear is generally for other applications, like measuring OD600). 3) Ensure that you have adequate controls. At the very least, have a nonfluorescent negative control of E. coli in the same media grown under the same conditions, either empty vector or isolated from a colony that had lost its fluorescence. 4) Turn on the plate reader - there is a power button in the back next to the power cord - and then turn on the Magellan software on the computer next to it. Not doing it in this order may cause the software to fail to recognize the machine. 5) Select the option that says "raw data" and eventually you will find yourself at a configuration screen. 6) First, select the brand and type of your plate next to the "Plate definition" dropdown menu. 7) Second, using ctrl click, select the wells you want to measure. 8) Third, you must add a measurement step. Double click "Fluorescence Intensity." Set your Excitation and Emission values to integers appropriate for your fluorophore and change Gain to "Optimal". You can also manually set gain by testing different values: generally, a higher gain detects smaller amounts of fluorescence; if you get overflow reads in several wells, you should set the gain lower. 9) Fourth, perform the measurements and save your data. Unfortunately, there is no Excel installed on that computer, so "Copy to Excel" is not a possibility at this point. Data is saved by default as ".wsp" files. (Still looking for a workaround - there is also a "Copy as ASCII" option that I haven't tested yet). 10) Finally, after performing this over a number of days, calculate how RFU changes as a function of time. Remember to normalize RFU to OD600, as a higher number of cells will naturally fluoresce more brightly than a lower number of cells given that both of the populations have the same proportion of fluorescent cells. | |||||||
| > > |
| |||||||
| Changed: | ||||||||
| < < | Troubleshooting: a) If you are not sure what the excitation/emission values for your fluorophore are, here is a handy reference courtesy of Simon: Spectra of Common Fluorophores. b) You can BLAST the gene sequence of your fluorophore to check which it matches (most likely a variant of EGFP). If you are still not sure or getting bizarre results, you may want to do a "Fluorescence Intensity Scan" where you set the plate reader to test numerous wavelengths of excitation in a certain range and then look at the spectra to see where the peaks (highest RFUs) are, in terms of wavelength. | |||||||
| > > | Troubleshooting:
| |||||||
Measurement by Flow CytometryNote
Measurement by Next-Gen SequencingExpected Results | ||||||||
Revision 42016-03-22 - LucyLeblanc
Evolutionary Stability of Fluorescent Protein ExpressionProcedure: Growth
Procedure: Measurement Measurement by Plating
There are essentially two ways to do this by plating, either by spreading an inoculum across the whole plate or by spotting inocula as serial dilutions. Both involve counting colonies and the readout is the ratio of fluorescent colonies divided by the number of total colonies. Though spreading consumes a significantly higher number of plates, it also allows detection of smaller changes in fluorescence, as typically 30-300 colonies can be counted on such plates while only about 6-60 colonies can be counted during spotting. | ||||||||
| Changed: | ||||||||
| < < | 10) Finally, after performing this over a number of days, calculate how RFU changes as a function of time. | |||||||
| > > | 10) Finally, after performing this over a number of days, calculate how RFU changes as a function of time. Remember to normalize RFU to OD600, as a higher number of cells will naturally fluoresce more brightly than a lower number of cells given that both of the populations have the same proportion of fluorescent cells. | |||||||
|
Troubleshooting: a) If you are not sure what the excitation/emission values for your fluorophore are, here is a handy reference courtesy of Simon: Spectra of Common Fluorophores. b) You can BLAST the gene sequence of your fluorophore to check which it matches (most likely a variant of EGFP). If you are still not sure or getting bizarre results, you may want to do a "Fluorescence Intensity Scan" where you set the plate reader to test numerous wavelengths of excitation in a certain range and then look at the spectra to see where the peaks (highest RFUs) are, in terms of wavelength. Measurement by Flow CytometryNote
Measurement by Next-Gen SequencingExpected Results | ||||||||
Revision 32016-03-21 - LucyLeblanc
Evolutionary Stability of Fluorescent Protein ExpressionProcedure: Growth
Procedure: Measurement | ||||||||
| Changed: | ||||||||
| < < | Measurement by Plating | |||||||
| > > | Measurement by Plating | |||||||
| Added: | ||||||||
| > > | There are essentially two ways to do this by plating, either by spreading an inoculum across the whole plate or by spotting inocula as serial dilutions. Both involve counting colonies and the readout is the ratio of fluorescent colonies divided by the number of total colonies. Though spreading consumes a significantly higher number of plates, it also allows detection of smaller changes in fluorescence, as typically 30-300 colonies can be counted on such plates while only about 6-60 colonies can be counted during spotting. | |||||||
| Changed: | ||||||||
| < < | ||||||||
| > > | Spreading | |||||||
| Added: | ||||||||
| > > | 1) Estimate the cfu/ml of the overnight culture via OD600 measurement. There is a web calculator available here. Note that to measure OD600 accurately you may have to dilute your culture as this is most accurate in the range of OD600 in the range of 0.1 to 1.0. 2) After estimation, dilute the culture in saline by taking 100 μL, adding it to 10 mL of saline, vortexing, and repeating the dilution process as needed. The ultimate goal is to dilute the cfu/ml to a point where spreading 50-100 μL would yield about 30-300 colonies. 3) Add sterile glass beads to LB agar plates with antibiotic, then place the appropriate amount of the diluted culture on the beads. 4) Shake the plates back and forth to ensure that the inoculum is spread uniformly across the surface of the agar. 5) Incubate at 37°C for 24 h. 6) Count the total number of colonies overall. If desired, you may use a colony-counting software such as OpenCFU, but it may be easier to simply do it by hand. If you do decide to use such software, please double-check the automated counts to eliminate any false positives and false negatives. 7) Count the total number of nonfluorescent colonies using blue light. Remember to place the orange filter on top and, if desired, wear the orange safety glasses. 8) Now, to calculate the ratio, subtract the number of nonfluorescent colonies from the number of total colonies. This will yield the number of fluorescent colonies. Then divide by the total number of colonies. 9) Plot the ratio as a function of time over several days. The expected result is a significant drop in this ratio over the course of a few days representing the decay of the genetic device. Spotting Note: This protocol was adapted from Thomas, Sekhar, Upreti, Mujawar, & Pasha (2015). A convenient flowchart of the protocol (though altered) can be found here. 1) It is no longer necessary to estimate the cfu/ml of the overnight culture, as each plate is capable of enumerating colonies over a range of 6 orders of magnitude as opposed to the above method. Instead of diluting the overnight culture in 10 ml of saline, set up a serial dilution series of sterile microfuge tubes each containing 900 μl of saline. Each tube represents a tenfold dilution (and thus should be labeled -1, -2, -3, etc.) and I recommend diluting out to 10^-7. Contrary to what the paper advises, I do not recommend diluting the overnight culture to an OD600 of 0.1; it is unnecessary. 2) Place 100 μl of the overnight culture into the microfuge tube representing 10^-1 and vortex. Please note that taking 100 μl from this would actually be a 10^-2 dilution (the overnight culture is first diluted tenfold after being added to the saline, then you only take 1/10th of the dilution to plate). This will be accounted for in the final formula to back calculate cfu/ml (if desired). 3) Label your LB agar plates with antibiotic by dividing most of the plate into 6 sections. Each section of the plate will be used to spot a different dilution of the same culture. Thus, the six sections may be labeled -2, -3, -4, -5, -6, and -7, for example. 4) Spot 10 μl of each dilution in triplicate onto the corresponding section of the plate. The paper recommends 20 ul but that seems to cause the spots to run into one another and it also takes longer to dry, increasing the chance for contamination. 5) Dry out the plate in the 30°C or 37°C incubator with the lid slightly off to allow moisture to evaporate. This part can take 15-20 mins depending on temperature and how moist the agar itself is. 6) Place the lid back on fully and incubate at 37°C for 24 h. The resulting plate should look like the example plate on the left here. 7) Calculate the ratio of fluorescent to nonfluorescent colonies. 8) Plot the ratio as a function of time over several days. Troubleshooting: a) The incubation period is around 24 h because colonies that are too small may not appear fluorescent (even if the genetic device is intact) and thus act as false negatives. However, growth rates can vary and you should check on colony growth before 24 h if colonies have grown too large and start to overlap. b) Fluorescent colonies can stay fluorescent for a couple of weeks in the fridge, so feel free to store them at 4°C and count them later if desired. c) If there is large variation between technical replicates, ensure you are vortexing for a few seconds after every dilution step. d) While drying the inocula during the spotting method, even though the plate is somewhat open and exposed for a period of time, it is unlikely that you will get any contamination. If this turns out to be a problem, you may also dry your plates in a biosafety cabinet which will take longer due to lower temperature but reduce the risk of contamination. | |||||||
Measurement by Microplate Reader1) Plan out which wells will contain what sample. You can print out one of the 96 well plate templates available online. Generally, each condition should be done in quadruplicate (or triplicate depending on space concerns).2) After vortexing your sample, aliquot 100μL at a time into each well. You should use the appropriate type of plate for your application: black walled, clear bottom for fluorescence (white walled, clear bottom is generally for luminescence and completely clear is generally for other applications, like measuring OD600). 3) Ensure that you have adequate controls. At the very least, have a nonfluorescent negative control of E. coli in the same media grown under the same conditions, either empty vector or isolated from a colony that had lost its fluorescence. 4) Turn on the plate reader - there is a power button in the back next to the power cord - and then turn on the Magellan software on the computer next to it. Not doing it in this order may cause the software to fail to recognize the machine. 5) Select the option that says "raw data" and eventually you will find yourself at a configuration screen. 6) First, select the brand and type of your plate next to the "Plate definition" dropdown menu. 7) Second, using ctrl click, select the wells you want to measure. 8) Third, you must add a measurement step. Double click "Fluorescence Intensity." Set your Excitation and Emission values to integers appropriate for your fluorophore and change Gain to "Optimal". You can also manually set gain by testing different values: generally, a higher gain detects smaller amounts of fluorescence; if you get overflow reads in several wells, you should set the gain lower. 9) Fourth, perform the measurements and save your data. Unfortunately, there is no Excel installed on that computer, so "Copy to Excel" is not a possibility at this point. Data is saved by default as ".wsp" files. (Still looking for a workaround - there is also a "Copy as ASCII" option that I haven't tested yet). 10) Finally, after performing this over a number of days, calculate how RFU changes as a function of time. Troubleshooting: a) If you are not sure what the excitation/emission values for your fluorophore are, here is a handy reference courtesy of Simon: Spectra of Common Fluorophores. b) You can BLAST the gene sequence of your fluorophore to check which it matches (most likely a variant of EGFP). If you are still not sure or getting bizarre results, you may want to do a "Fluorescence Intensity Scan" where you set the plate reader to test numerous wavelengths of excitation in a certain range and then look at the spectra to see where the peaks (highest RFUs) are, in terms of wavelength. Measurement by Flow CytometryNote
Measurement by Next-Gen SequencingExpected Results | ||||||||
Revision 22016-03-04 - LucyLeblanc
Evolutionary Stability of Fluorescent Protein ExpressionProcedure: Growth
Procedure: MeasurementMeasurement by PlatingMeasurement by Microplate Reader | ||||||||
| Changed: | ||||||||
| < < | ||||||||
| > > | 1) Plan out which wells will contain what sample. You can print out one of the 96 well plate templates available online. Generally, each condition should be done in quadruplicate (or triplicate depending on space concerns). | |||||||
| Added: | ||||||||
| > > | 2) After vortexing your sample, aliquot 100μL at a time into each well. You should use the appropriate type of plate for your application: black walled, clear bottom for fluorescence (white walled, clear bottom is generally for luminescence and completely clear is generally for other applications, like measuring OD600). 3) Ensure that you have adequate controls. At the very least, have a nonfluorescent negative control of E. coli in the same media grown under the same conditions, either empty vector or isolated from a colony that had lost its fluorescence. 4) Turn on the plate reader - there is a power button in the back next to the power cord - and then turn on the Magellan software on the computer next to it. Not doing it in this order may cause the software to fail to recognize the machine. 5) Select the option that says "raw data" and eventually you will find yourself at a configuration screen. 6) First, select the brand and type of your plate next to the "Plate definition" dropdown menu. 7) Second, using ctrl click, select the wells you want to measure. 8) Third, you must add a measurement step. Double click "Fluorescence Intensity." Set your Excitation and Emission values to integers appropriate for your fluorophore and change Gain to "Optimal". You can also manually set gain by testing different values: generally, a higher gain detects smaller amounts of fluorescence; if you get overflow reads in several wells, you should set the gain lower. 9) Fourth, perform the measurements and save your data. Unfortunately, there is no Excel installed on that computer, so "Copy to Excel" is not a possibility at this point. Data is saved by default as ".wsp" files. (Still looking for a workaround - there is also a "Copy as ASCII" option that I haven't tested yet). 10) Finally, after performing this over a number of days, calculate how RFU changes as a function of time. Troubleshooting: a) If you are not sure what the excitation/emission values for your fluorophore are, here is a handy reference courtesy of Simon: Spectra of Common Fluorophores. b) You can BLAST the gene sequence of your fluorophore to check which it matches (most likely a variant of EGFP). If you are still not sure or getting bizarre results, you may want to do a "Fluorescence Intensity Scan" where you set the plate reader to test numerous wavelengths of excitation in a certain range and then look at the spectra to see where the peaks (highest RFUs) are, in terms of wavelength. | |||||||
Measurement by Flow CytometryNote
Measurement by Next-Gen SequencingExpected Results | ||||||||
Revision 12016-02-23 - JeffreyBarrick
Evolutionary Stability of Fluorescent Protein ExpressionProcedure: Growth
Procedure: MeasurementMeasurement by PlatingMeasurement by Microplate ReaderMeasurement by Flow CytometryNote
Measurement by Next-Gen SequencingExpected Results |
View topic | History: r11 < r10 < r9 < r8 | More topic actions...