RNAseq Library Preparation
Use RNA from RNASnap and Zymo column purification as outlined on wiki.rRNA Depletion
Use the Ribozero (RZ) rRNA Removal Kit (Gram-negative bacteria) from Illumina. We assayed the effectiveness of the RZ kit: using inputs of up to 5ug, rRNA was reduced from >80% of total cell RNA to less than 1%. Protocol as follows: *Cool a benchtop centrifuge to 40C, place 100% ethanol and 70% ethanol on ice (store chilled at -200C). Set a waterbath to 500C* DO NOT PUT MAGNETIC BEADS ON ICE. All RZ components can be kept at room temperature during the procedure. Protocol: Batch wash magnetic beads:- Allow beads to equilibrate at RT.
- Vortex beads to homogeneity.
- Pipette required volume of beads for #reactions - 225ul of beads / reaction.
- Place tube containing correct volume of beads on magnetic stand for 1 minute til clear.
- Remove supernatant.
- Remove tube from stand, add equal (to original bead suspension) volume of R.F. H2O.
- Vortex vigorously.
- Repeat wash.
- Remove wash H2O, add 60ul Resuspension Solution and 1ul Riboguard / reaction.
- Vortex.
- Aliquot 65ul of beads to new individual 1.5ml tubes / reaction.
- Vortex.
- Store at RT.
- Allow kit components to equilibrate to RT
- Per sample, add following individually and in order, in a PCR strip tube:
| R.F. H2O |
X |
| rRNA reaction buffer |
4 |
| RNA sample |
Y (up to 5ug, lower limit not assayed) |
| Removal Solution |
10 |
| TOTAL |
40 |
- Mix by pipetting 10-15x.
- Incubate @ 68°c, 10 mins.
- Centrifuge briefly to collect condensation.
- Incubate @ RT, 5 mins.
- Centrifuge briefly to collect condensation.
- Transfer hybridized reaction to magnetic beads ( not the other way round), and immediately and thoroughly mix by pipetting 10-15x.
- Vortex at high speed for 15 seconds.
- Incubate tubes at RT, 5mins.
- 50°c, 5 mins.
- Immediately place tubes on mag stand and allow to clear.
- Remove supernatant (80-85ul) and store on ice.
ETOH precipitation purification
The pellets - including the final pellet - may not be easily visible, so keep track of which 'corner' of the centrifuge tube is facing outwards in centrifuge. Protocol:- Adjust samples to 180ul w/ R.F. H2O
- +18ul 3M Na acetate, pH 5.2
- +2ul glycogen, 10mg/ml
- +600ul ice cold 100% ethanol
- vortex gently
- -80°c, 30mins; -20°c, 30mins.
- C’fuge @ >10K x g 30 mins, 4°c.
- Remove supernatant, wash pellet with ice cold 70% ETOH.
- C’fuge @ 10K x g for 5 mins, 4°c.
- Repeat 70% wash.
- Remove all 70%, first with p1000 then with p20. Invert tube on to parafilm and air dry for 5 mins.
- Resuspend in 18ul R.F. H2O and immediately transfer to PCR strip tube on ice.
RNA Fragmentation
Use NEB Fragmentation Buffer. The RNA frag buffer conditions and high temperature cause RNA fragmentation. Fragment size is determined by time exposed to both. For tight control of fragmentation, add the buffer to the RNA in a strip tube, seal, resuspend tube by thoroughly flicking strip, centrifuge briefly and place in a pre-heated PCR cycler. Likewise, at the end of the run immediately cool on ice, collect any condensation by centrifugation and then add Stop Buffer. For E.coli, in our hands, 2 minutes at 94 degrees produces a final PCR product smear (see final steps) of between 150bp and 500bp (>98% of product between 150bp and 300bp). This should be determined experimentally from lab to lab. Protocol:- Mix the following components in a sterile PCR tube, in order:
| RNA (above) |
18 ul |
| RNA Frag buffer (10x) |
2 ul |
| Total volume |
20 ul |
- Incubate in a preheated thermal cycler for 1-5 minutes (see above) at 94°c.
- Transfer tube to ice immediately.
- Centrifuge to collect condensation. Add 2 ul of 10x RNA Fragmentation stop solution.
- Ethanol precipitate as for above (possible to reduce precipitation volumes to save on sodium acetate, glycogen etc if necessary).
- During the final 5 minute drying phase, make up the master mix for T4 kinase treatment and aliquot to n empty PCR strip tube wells (on ice). Cover with parafilm.
- Resuspend final RNA pellet in 14ul of H2O and immediately transfer to PCR strip tube on ice containing the reaction buffer for T4 kinase treatment (below).
Kinase Treatment of Fragmented mRNA
A master mix of buffer, T4 PNK and ATP can be made and 6ul added to n empty PCR strip tube wells (on ice) during the above ethanol precipitation's final 5 minute drying phase (see note above). Then resuspended RNA can be added to the reaction mix and thoroughly mixed immediately. Protocol:| Fragmented mRNA (above) |
14ul |
| T4 PNK buffer, 10x |
2.0 ul |
| T4 PNK |
2.0 ul |
| ATP, 10 mM |
2.0 ul |
| Total Volume per rxn |
20.0 ul |
- Mix contents of tubes.
- Incubate at 37°c for 30 minutes.
- Snap-cool on ice.
- Ethanol precipitate using above exact method OR precipitate overnight at -20°c, pelleting the next morning as usual.
- NB: This is the last pause point before PCR amplification, steps after this must be done same day!
- Resuspend the final pellet in 6ul of H2O, and transfer immediately to a PCR strip tube.
Library Preparation
Use the NEBNext Small RNA Library Prep Set for Illumina (Multiplex Compatible), catalogue #E7330S/L (24/96 rxns).
The entire protocol is carried out in a single tube per sample, each subsequent reaction mixture being added to the previous product. Care should therefore be taken after each step to cool the products on ice and collect them by brief centrifugation so as to maintain reaction volume.
Note that many of the below steps require pre-heated blocks or transfers between temperatures. Plan accordingly.
Due to the low concentration of RNA following RZ, we routinely dilute the adapters, primers etc that come in the kit 1 in 2 (e.g. 1ul H2O + 1ul primer)
Protocol:
3’ SR Adaptor Ligation - Add 1ul of diluted 3' SR adaptor to each 6ul RNA sample prepared above, seal, flick thoroughly to mix, centrifuge to collect.
- Incubate in a preheated thermal cycler for 2 minutes at 700C.
- Transfer tube to ice, collect by centrifugation.
- Add the following, separately and in order(given that both the buffer and the enzyme mix are viscous, we find it easier to handle accurate additions separately).
| 3' ligation buffer | 10ul |
| 3' ligation enzyme mix | 3ul |
| Total cumulative reaction volume | 20ul |
- Seal, flick thoroughly to mix, collect by centrifugation.
- Incubate for 1 hour at 250C (do not use heated lid).
- Add the following to each reaction from step 1:
| H2O | 4.5ul |
| Diluted SR RT primer | 1ul |
| Previous reaction product | 20ul |
| Total cumulative reaction volume | 25.5ul |
- Heat sample (3 different times and temperatures):
-
- 5 min at 750C (heated lid at 800C)
- 15 min at 370C (heated lid at 420C)
- 15 min at 250C (do not use heated lid). During this incubation, proceed to 'denature 5' SR adapter' below.
- Centrifuge to collect any condensation.
- Denature 5' SR adapter: With 5 minutes remaining in step C above, aliquot 1.1n ul of the 5’ adaptor into a PCR strip tube (where n = number of samples being processed).
- Incubate the 5’ adapter in a preheated thermal cycler at 700C for 2 minutes.
- Immediately place the tube on ice and use within 30 minutes.
- Add the following, individually, to the prepared RNA from steps above:
| Diluted 5’ SR adapter (denatured as above) | 1ul |
| 5’ ligation reaction buffer (10x) | 1ul |
| 5’ ligation enzyme mix |
2.5ul |
| Previous reaction product | 25.5ul |
| Total cumulative reaction volume | 30ul |
- Seal, flick thoroughly to mix, collect by centrifugation.
- Incubate for 1 hour at 250C (do not use heated lid).
- Make a stock of buffer, RNase inhibitor and RTase and use it to add the following to each rxn:
| First strand synthesis buffer | 8ul |
| Murine RNase inhibitor | 1ul |
| M-MuLV Reverse Transcriptase (RNase H-) | 1ul |
| Previous reaction product | 30ul |
| Total cumulative reaction volume | 40ul |
- Incubate for 1 hour at 500C.
- Cool on ice, collect condensation by centrifugation.
- Add the following to the RT reaction mix from step 4 (make a master mix of LongAmp Taq mix, diluted SR primer, and nuclease free water; add the Index primer individually).
| LongAmp Taq 2x Master mix | 50ul |
| Diluted SR primer | 2.5ul |
| Nuclease free water | 5.0ul |
| Inidividually added Index primer | 2.5ul |
| Previous reaction product | 40ul |
| Total volume | 100ul |
- Mix thoroughly by pipetting
- In some PCR cyclers, it may be necessary to split the reaction volume in to two separate 50ul reactions for efficient heating/cooling.
| Cycle Step |
TEMP |
Time |
Cycles |
| Initial denaturation |
940 C |
30 sec |
1 |
| Denaturation |
940 C |
15 sec |
12-15 |
| Annealing |
620 C |
30 sec |
12-15 |
| Extension |
700 C |
15 sec |
12-15 |
| Final Extension |
700 C |
5 min |
1 |
| Hold |
40 C |
- |
|
- Ethanol precipitate as above (pellet should be large).
- Dissolve the pellet in 20ul of TE buffer or ddH20.
- QC: Qubit (check ng/ul)
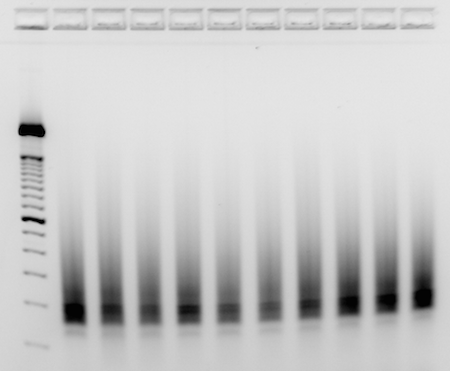
- Load 200ng of finished library into each well of a 4% EX E-gel, with 200ng of 50bp ladder as reference
- Run until smear is obvious from 200bp+, and products can be easily resolved from any undesirable amplicons at lower sizes.
- Take picture of gel with imager
- Open gel
- Excise bands from 150bp to the top of the ladder using a clean razor blade for each lane.
- Gel purify using Zymo gel recovery kit (use 550 C during gel dissolution step).
- Elute in 50-100ul H2O.
- Qubit for DNA concentration, submit to GSAF as per their recommendations.
- Example_RNAseq_libraries:


| I | Attachment | History | Action | Size | Date | Who | Comment |
|---|---|---|---|---|---|---|---|
| |
Barrick_Lab_2016-01-27_RNAlib_2.png | r1 | manage | 131.9 K | 2017-01-30 - 19:28 | UnknownUser | Example_RNAseq_library |
{kind=link}
{kind=link}
Barrick Lab > ProtocolList > RNASeq