Golden Gate Assembly: Creating a New Part
This protocol describes how to clone a new Golden Gate part amplified from a template DNA (like a plasmid or a genome) or synthesized as a piece of double-stranded DNA (like an IDT gBlock) into the YTK001 entry vector via a BsmBI assembly reaction.Supplies
- dsDNA piece encoding the part with restriction sites appended (see below)
- Phusion® High-Fidelity DNA Polymerase (NEB: M0530)
- 10× T4 DNA ligase buffer (NEB: M0202)
- T7 DNA ligase (NEB: M0318)
- Restriction endonuclease BsmBI (NEB: R0580)
- Competent cells
- SOC and liquid media for recovery after transformation
- LB+Cam plates for recovering transformants with assembled plasmid
Step 1: Creating dsDNA encoding the part for cloning
Two variations on preparing a dsDNA fragment with the proper restriction sites and Golden Gate overhangs are provided in the next sections. The end result is the same: a piece of DNA with the proper flanking regions for BsmBI cloning into the entry vector, while maintaining the BsaI sites used in first stage assembly – with proper overhangs for the type of part that you are designing.Method 1: Synthesizing dsDNA containing the required flanking regions
When ordering a double-stranded piece of DNA to be synthesized, you can just append the sites needed for cloning in your order. Use this protocol when ordering a gBlock from IDT, for example. 5'-GCATCGTCTCATCGGTCTCAXXXX YOUR PART YYYYTGAGACCTGAGACGGCAT-3'3'-CGTAGCAGAGTAGCCAGAGTxxxx your part yyyyACTCTGGACTCTGCCGTA-5' Replace XXXX with the prefix-F sequence for the part type you are designing ( see the table and notes below). Similarly, replace YYYY with the suffix-F sequence for the part. In general, the amount of DNA synthesized is sufficient for cloning into the entry vector without further PCR amplification of a gBlock.
Method 2: Amplifying a sequence with primers that add the required flanking regions
You need to order two primers that append sequences to the ends of the DNA sequence that you are amplifying. If your template looks like the following, then you should design the two primers like the examples. Template 5'-UUUUUUUUUUUUUUUUUUUU YOUR PART DDDDDDDDDDDDDDDDDDD-3'3'-uuuuuuuuuuuuuuuuuuuu your part ddddddddddddddddddd-5' Primer 1 (forward) 5'-GCATCGTCTCATCGGTCTCAXXXXUUUUUUUUUUUUUUUUUUUU-3' Primer 2 (reverse) 5'-ATGCCGTCTCAGGTCTCAyyyyddddddddddddddddddd-3' CAUTION In Primer 2, the priming site (dddd...) must be the reverse-complement of your part and you must use the suffix-R sequence for the Golden Gate part overlap because this is on the other strand. The overlap with the template can vary from the 20 base pairs that are shown according to the normal rules for designing good PCR primers. If calculating melting temperatures, be sure to only include the overlap region in your calculations, not the stuff that is being added to the ends!
Example PCR Reaction
- PCR Insert
- Use standard 25ul Phusion (or other high fidelity polymerase) protocol
- PCR (25ul reaction)
- 5 ul 5x Buffer
- 1.5 ul dntps
- 1.25 ul primer (A+C)
- 1.25 ul primer (B+D)
- x ul template plasmid (<250ng)
Part Overlap Quick Reference Table
These part overhangs are used by the YTK (yeast toolkit) and the BTK (Broad-host-range toolkit). More information on the design of parts for Golden Gate assembly using these standards can be found in the reference to the YTK (especially in the supplement). Be aware that some definitions vary between the two toolkits.| Type | Description | Prefix (F) (XXXX) | Prefix (R) (xxxx) | Suffix (F) (YYYY) | Suffix (R) (yyyy) | Notes |
| 2 | promoter + RBS | AACG | cgtt | TATG | cata | |
| 3 | (BTK) gene | TATG | cata | ATCC | ggat | The start codon (ATG) is within the prefix. After that, begin your protein coding sequence in-frame with the second codon. BTK: Include the stop codon for your gene!. |
| 3 | (YTK) gene | TATG | cata | GGATCC | ggatcc | The start codon (ATG) is within the prefix. After that, begin your protein coding sequence in-frame with the second codon. YTK: Omit the stop codon. It is typically placed in the next part. For that stop codon to be in-frame, you must end this part with the two bases GG so that plus the suffix encode a Gly-Ser linker/extension. |
| 4 | (BTK) terminator | ATCC | ggat | GCTG | cagc | |
| 4 | (YTK) stop codon + terminator | ATCCTAA | ttaggat | GCTG | cagc | The first three bases within the part should be a stop codon (TAA). |
- All sequences in the table are written 5' to 3'.
- YTK: yeast toolkit; BTK: broad-host-range toolkit.
- Standard bases that are added before or after the actual Golden Gate overhang in prefix / suffix to maintain part function are shown in red.
Step 2: Golden Gate Assembly Reaction
Now, you perform a Golden Gate Assembly reaction with BsmBI to ligate your part into the entry vector (pYTK001).Below is the standard Barrick lab Golden Gate Assembly (GGA) reaction. Unless otherwise specified, you should use this setup. If you are in FRI/iGEM, consider using the alternative Step 2B procedure below. Total volume will be 20 μL; You will need 17.7 ng of pYTK001 and 20 fmol of your DNA insert(s).
- 17.7ng pYTK-001 plasmid [try to keep volume to 1-2μL]
- 20 fmol of insert DNA = 650 * insert length * 20x10^-6 ng [try to keep volume to less than 10 μL]
- 2 μL of 10× T4 DNA ligase buffer
- 1 μL of BsmBI
- 1 μL of T7 DNA ligase
- x μL water up to 20 μL total.
Mix well by pipetting.
- Incubate the reaction at (42°C for 5 min and then 16°C for 5 min) *30 , followed by 10 min at 55°C, 10min at 80 °C .
- Use 2 μL assembly reaction for electroporation.
- After recovery, plate on LB/CAM.
Step 2B: Alternative Golden Gate Assembly Reaction (FRI/iGEM variant)
The reason for this alternative procedure is based on some summer experiments, showing that one reason we may have high background is that we are using BsmBI at non-ideal conditions. This first gel shows pYTK001 being cut with BsmBI and pYTK095 being cut with BsaI at either 37°C or 42°C with either T4 DNA ligase buffer, cutsmart buffer, or NEB 3.1 buffer. This last buffer is the preferred BsmBI buffer. BsmBI is most active at 55°C (NEB catalog). All buffers and enzymes are from NEB (New England Biolabs)- Gel of BsmBI digestion under different reaction conditions.:
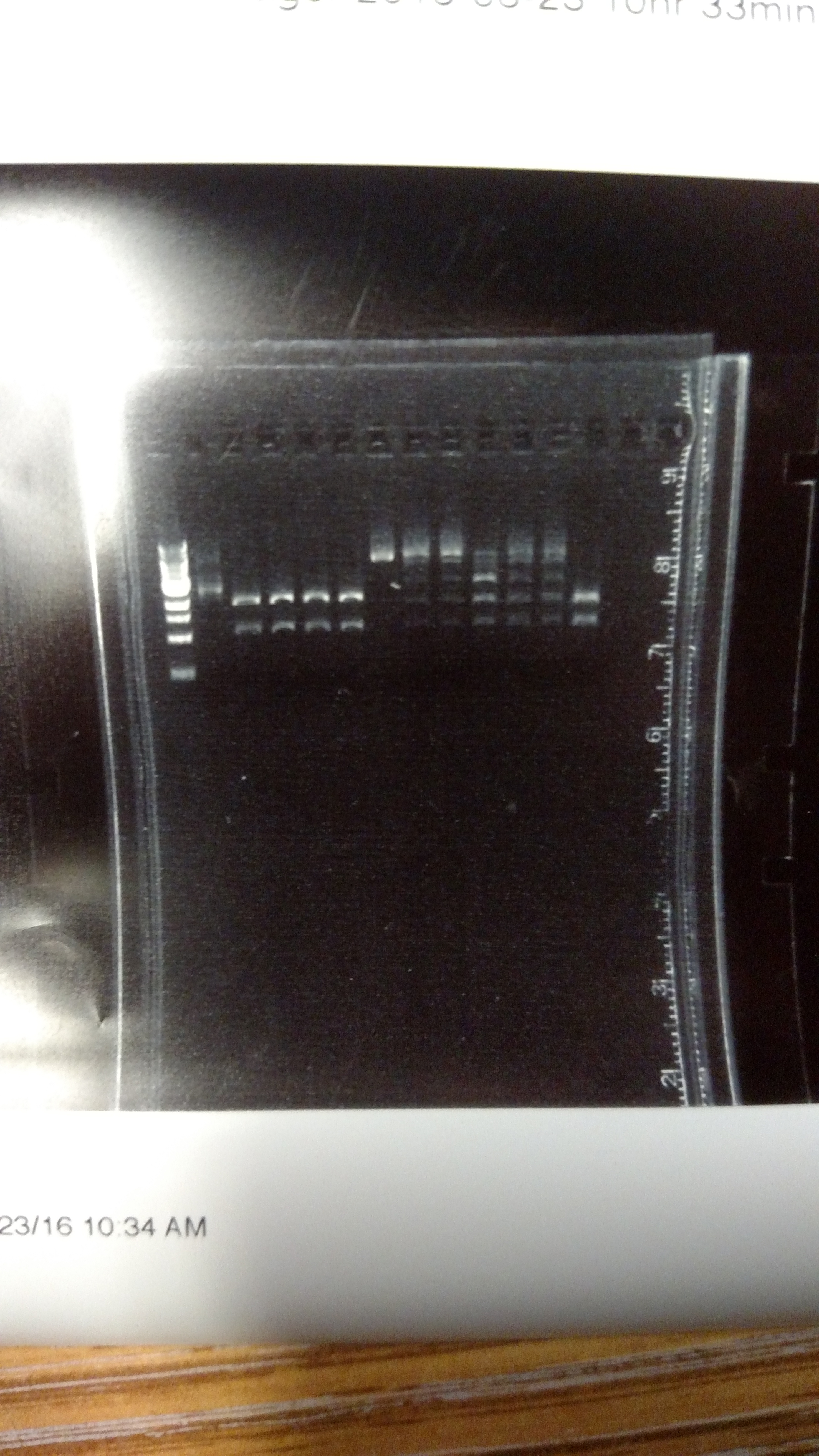
From left to right: (Note: Not sure how to get the text to move over a bit more...-DM)
- 1 KB ladder
- uncut pYTK095
- pYTK095 + cutsmart buffer at 37°C
- pYTK095 + T4 DNA ligase buffer at 37°C
- pYTK095 + cutsmart buffer at 42°C
- pYTK095 + T4 DNA ligase buffer at 42°C
- uncut pYTK001
- pYTK001 + cutsmart buffer at 37°C
- pYTK001 + T4 DNA ligase buffer at 37°C
- pYTK001 + 3.1 buffer at 37°C
- pYTK001 + cutsmart at 42°C
- pYTK001 + T4 DNA ligase buffer at 42°C
- pYTK001 + 3.1 buffer at 42°C
Based on the above reactions, a protocol was developed using NEB buffer 3.1, supplemented with ATP and DTT to allow for T7 DNA ligase activity. The results can be seen in the PDF below. There are three sets of reactions: The first lane for each set is DNA only, then DNA in a GGA reaction without ligase, and finally DNA in a GGA reaction with ligase. In each set the presence of the ligase results in the formation of higher band products. The procedure was to incubate for 30 minutes with BsmBI at 42°C followed by incubating with T7 DNA ligase at 25°C for 30 additional minutes.
- GGA_test2_6_23_2016.pdf: Test of BsmBI and T7 DNA ligase procedure using NEB buffer 3.1
This initial protocol was simply a proof of principle. From this protocol, we are currently using the following protocol. Total volume will be 20 μL; You will need 17.7 ng of pYTK001 and 20 fmol of your DNA insert(s).
- 17.7ng pYTK-001 plasmid [try to keep volume to 1-2μL]
- 20 fmol of insert DNA = 650 * insert length * 20x10^-6 ng [try to keep volume to less than 10 μL]
- 2 μL of 10× 3.1 buffer
- 2 μL of 10 mM ATP
- 1 μL of 100 mM DTT
- 1 μL of BsmBI
- x μL water up to 20 μL total.
- Incubate at 42°C for 30 minutes.
- Then, add 1 μL of T7 DNA ligase and incubate at 16°C for 30 minutes. This is followed by 25 cycles of incubation at 42°C for 5 minutes and 16°C for 5 minutes. This is then followed by a 15-30 minute incubation at 55°C and a 10 minute incubation at 80°C.
- Use 2 μL assembly reaction for electroporation.
- After recovery, plate on LB/CAM.
Results and Troubleshooting
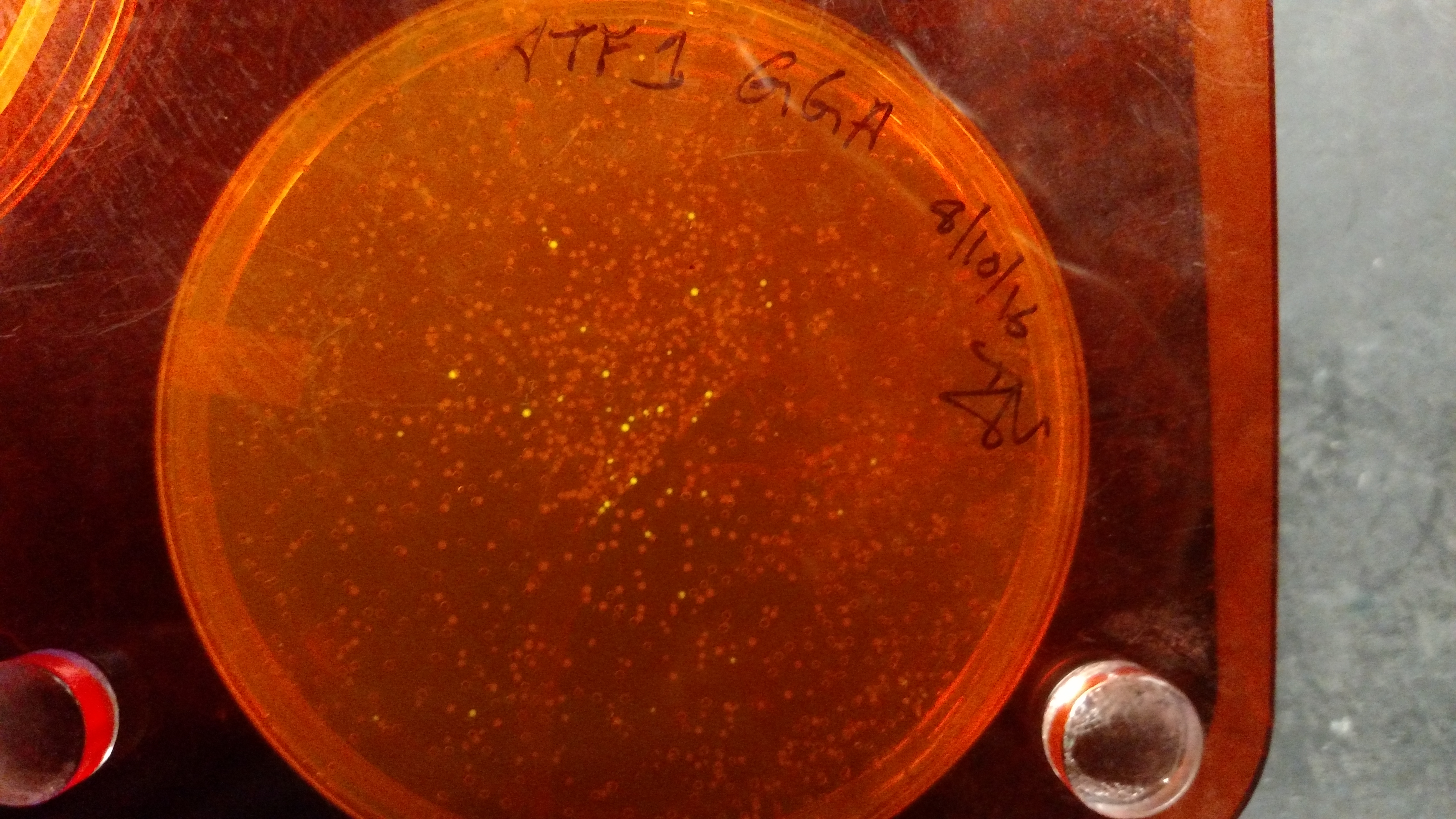
Below are three plates from summer 2016. ATF1 (bba_J45014) was added to pYTK001, an entry vector for golden gate assembly. This required removing an internal BsmBI cut site, and then adding the resulting ATF1 gene to pYTK001 using BsmBI. The first plate is a negative control. The golden gate assembly was performed with no gene, just pYTK001. The second plate is a positive control using a gBlock from Peng. The last plate is the actual experimental golden gate assembly where ATF1 inserted into pYTK001.- The Negative control GGA using only pYTK001 with no insert.:
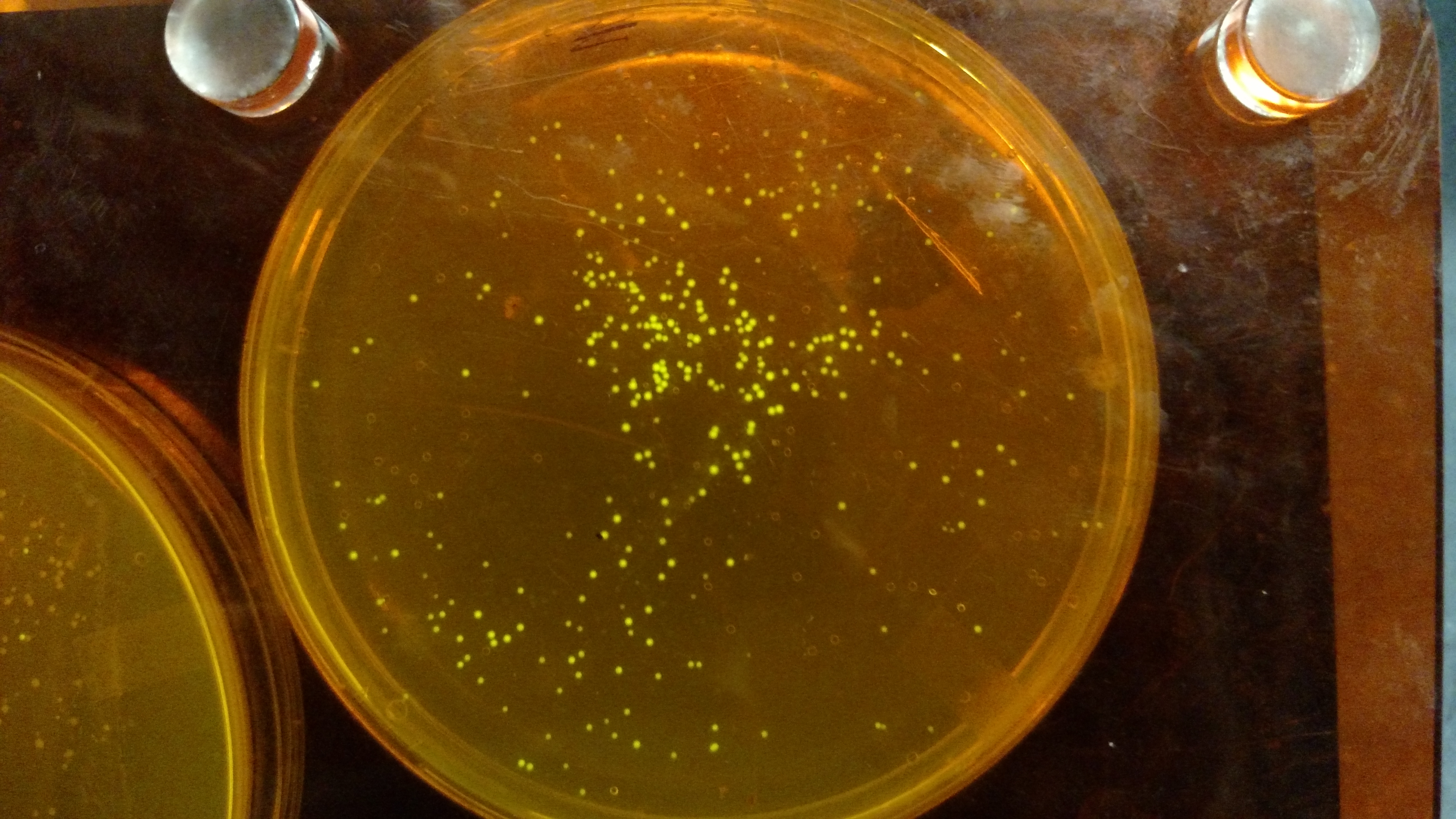
- The Positive control GGA using a gBlock from Peng with pYTK001.:
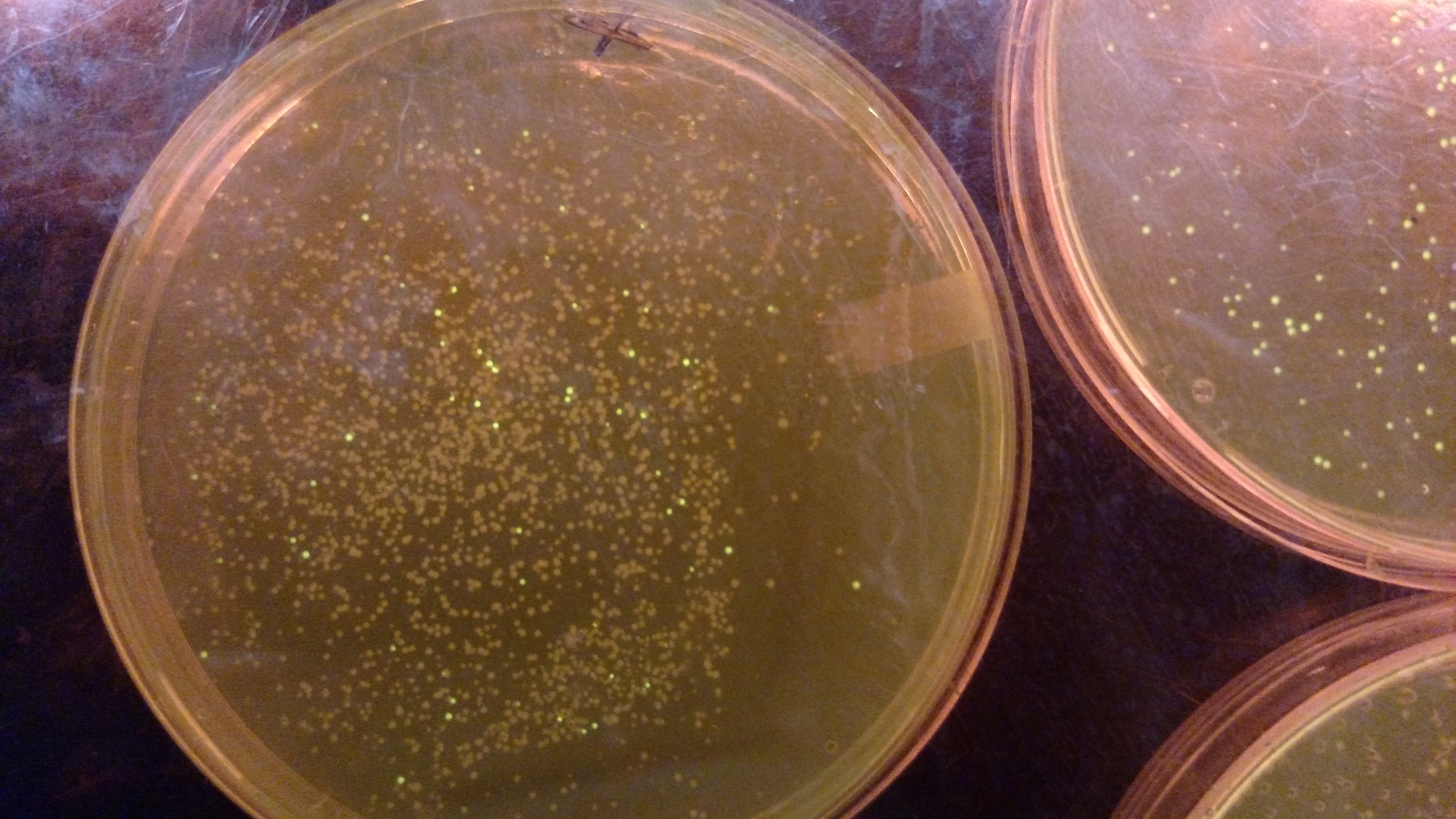
- ATF1-pYTK001 GGA transformation on LB/CAM plate.:
gBlocks vs PCR products as sequences being assembled.
- gBlocks - This high quality dsDNA tends to easily be assembled into an entry vector, such as pYTK001. This DNA can be resuspended and thrown straight into a GGA reaction.
- PCR products - This DNA can vary in quality, and tends to be more troublesome in two ways.
- Since the DNA is synthesized via PCR, there will frequently be point mutations. Thus, when picking colonies from a plate after GGA, try to pick at least 3 colonies. With ATF1-pYTK001 (see the plates above), 5 colonies were picked, 3 had point mutations.
- We have noticed that the PCR products made and purified in the FRI lab may be "less stable" or perhaps "degrades more quickly". It is unclear if this is a true difference, but regardless multiple PCR products were partially or fully degraded within 1 month of purification and storage at -20 degrees. Thus, it is STRONGLY recommended that immediately after purifying the PCR products, you should Qubit the products, and then begin your assembly reaction. Example: when creating an ATF1 PCR product that was compatible for golden gate assembly, the ATF1 gene first needed to be PCR'd into two discreet products to remove a restriction site. These two PCR products were then used in an GGA reaction with pYTK001 to create a single plasmid. However, the first couple of times this was attempted, the PCR products were made ~1 week before being used in the assembly reaction. These assemblies failed, and upon inspection (~1 week later), the PCR products were partially or wholly degraded. On the final attempt (which appears to have worked), the PCR products were purified on the same day that they were assembled into the pYTK001 entry vector. This is not conclusive, but it suggests that the quality of PCR products, which can naturally degrade over time (due to freeze/thaw cycles, nuclease contamination, or being left out at room temp), is absolutely critical. Even partial degradation could be sufficient to fully prevent successful GGA as the partially degraded products may still have one of their two cut sites available. These sites would be cleaved and could then be ligated to the pYTK001 plasmid. However, lacking a second viable cut site, this product would be an off pathway product (i.e. you can convert it into the desired final product).
References
- Lee ME, DeLoache WC, Cervantes B, Dueber JE. (2015) A Highly Characterized Yeast Toolkit for Modular, Multipart Assembly. ACS Synth. Biol. 4:975–986. Link
Contributors
- Peng Geng
- Jeffrey Barrick
- Dennis Mishler


| I | Attachment | History | Action | Size | Date | Who | Comment |
|---|---|---|---|---|---|---|---|
| |
GGA_test2_6_23_2016.pdf | r1 | manage | 1403.5 K | 2016-08-15 - 17:33 | DennisMishler | Test of BsmBI and T7 DNA ligase procedure using NEB buffer 3.1 |
| |
IMG_20160623_115229529.jpg | r1 | manage | 3864.3 K | 2016-08-15 - 17:35 | DennisMishler | Gel of BsmB I digestion under different reaction conditions. |
| |
IMG_20160811_080306611.jpg | r1 | manage | 4584.3 K | 2016-08-15 - 15:29 | DennisMishler | ATF1-pYTK001 GGA transformation on LB/CAM plate |
| |
IMG_20160811_080312832.jpg | r1 | manage | 4741.6 K | 2016-08-15 - 15:51 | DennisMishler | The Positive control GGA using a gBlock from Peng. |
| |
IMG_20160811_080319246.jpg | r1 | manage | 4508.7 K | 2016-08-15 - 15:51 | DennisMishler | The Negative control GGA using only pYTK001 with no insert. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Barrick Lab > ProtocolList > ProtocolsGoldenGateMakingANewPart
Topic revision: r16 - 2018-01-29 - 19:04:50 - Main.KateElston