Acinetobacter baylyi ADP1 Genome Manipulations by Overlap Extension PCR
Overlap PCR Construct Generation
The following is a standard procedure designing and constructing ADP1 genome manipulation constructs using overlap extension PCR. Amplified PCR products include annealing ends that allow them to bind to adjacent other targeted products for extension by an additional round of PCR.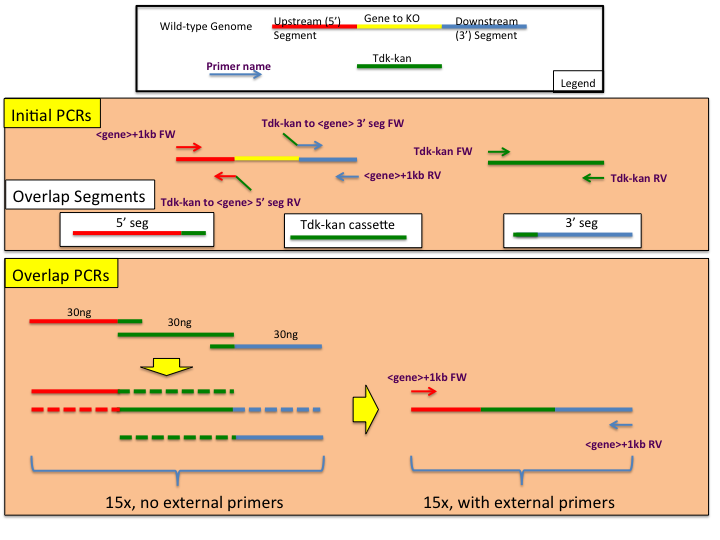
The Design of Overlap PCR Primers and Constructs
In order to ensure a successful assembly of transforming constructs, particular care should be made when designing the appropriate primers. A total of 6 primers are needed (see Initial PCRs panel in the provided figure), two of which will add homology of the tdk-kan cassette to the upstream flanking region (5' seg) and downstream flanking region (3' seg). Follow our dedicated page on primer design using Phusion polymerase where relevant. The following additional guidelines for primer construction should be followed:- Each homologous overlap should be at least 20bp long as well as each non-homology adding primer.
- When designing the overlap (or homology adding) primer, calculate the annealing temperature of each side of the primer separately. For example, if you have a primer NNNNNNNNNNNNNNYYYYYYYYYYYYYY where the Ns are homologous to the 3' end of the tdk-kan cassette and the Ys are homologous to the 5' end of the 3' seg piece, use only the Ys when calculating the annealing temperature for the 3' seg construction.
- The annealing temperature primers for the 5' seg and 3' seg amplification (green and red arrows in the diagram) should be within 2 degrees of each other - generally a Phusion Tm of approximately 62C is sought.
- Check the homologous overlap regions for hairpin formation using http://www.idtdna.com/analyzer/applications/oligoanalyzer/ . Stable hairpins at or above reaction annealing conditions will inhibit the overlap reaction.
Overlap PCR Reactions
Initial PCR of 5' Segment
In a PCR tube, combine the following:- 10uL 5X Phusion HF Buffer
- 1uL dNTP stock
- 2.5uL 10uM
+1kb FW primer - 2.5uL 10uM Tdk-kan to
5' seg RV primer - ~10ng wild-type ADP1 gDNA
- 0.3uL Phusion polymerase
- dH2O to 50uL
Initial PCR of tdk-kan Segment
In a PCR tube, combine the following:- 10uL 5X Phusion HF Buffer
- 1uL dNTP stock
- 2.5uL 10uM tdk-kan FW primer
- 2.5uL 10uM tdk-kan RV primer
- ~10ng ADP1 Knock out collection clone gDNA
- 0.3uL Phusion polymerase
- dH2O to 50uL
Initial PCR of 3' Segment
In a PCR tube, combine the following:- 10uL 5X Phusion HF Buffer
- 1uL dNTP stock
- 2.5uL 10uM Tdk-kan to
3' seg FW primer - 2.5uL 10uM
+1kb RV primer - ~10ng wild-type ADP1 gDNA
- 0.3uL Phusion polymerase
- dH2O to 50uL
Overlap PCR Reaction
In a PCR tube, combine the following:- 10uL 5X Phusion HF Buffer
- 1uL dNTP stock
- ~30ng 5'seg PCR
- ~30ng tdk-kan PCR
- ~30ng 3'seg PCR
- 0.3uL Phusion polymerase
- dH2O to 45uL
- 2.5uL 10uM
+1kb FW primer - 2.5uL 10uM
+1kb RV primer
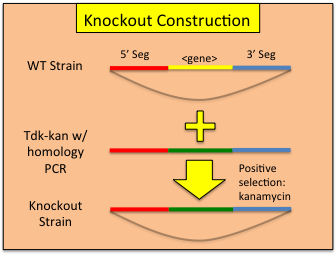
Constructing Gene Knockouts (or interruptions) in A. baylyi ADP1
A collection of single gene knockouts has been published ( http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2290942/ ) is currently available at http://www.genoscope.cns.fr/spip/Strain-request-for-mutants-of,749.html . However, if you wish to construct your own knockouts, use the following work flow:- Identify the region in which you would like to insert the tdk-kan cassette.
- Design and assemble your overlap construct (see above) such that homologous recombination with the genome will insert the construct at your desired location. One can either interrupt via insertion or replace the targeted gene to create a functional knock out.
- Transform wild-type ADP1 with your reserved 45uL of overlap assembly PCR as per our standard transformation protocol. Allow to grow at 30C overnight in a shaking incubator. Be sure to include a no-DNA transformation control.
- Plate both 100uL of undiluted and 100uL of a 1:100 dilution transformation onto dry LB-kan plates. Additionally, plate 100uL of undiluted control bacteria also onto a LB-kan plate. 1:1,000,000 dilutions for each reaction should also be plated on LB plates to allow transformation frequency to be calculated. Allow the plates to grow overnight in a 30C incubator.
- Pick 3 colonies from one of the transformation plates into 5mL liquid LB+kan each and allow to grow to confluence.
- Test each culture for the insertion via PCR and make a glycerol stock of successful insertions. PCR can be performed directly from the culture by diluting 1uL of culture into 1mL sterile dH2O and using 1uL of this as a PCR template. In the PCR reaction, the first 98C denaturing step should be changed from 30 seconds to 10 minutes to facilitate liberation of genomic DNA template.
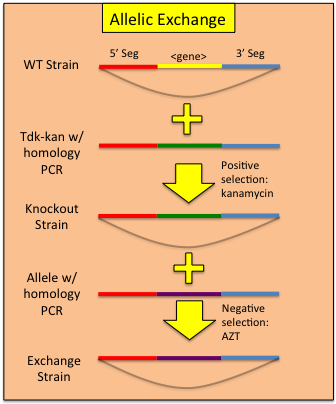
Constructing Allelic Replacements
Once one has replaced a gene in the ADP1 genome with the tdk-kan cassette, a mutated allele can be used to 'rescue' the bacteria from the cassette via transformation and plating on AZT. Note: zidovudine is the pharmaceutical name for AZT and can be ordered at far less cost. In our hands, we find zidovudine to be just as effective in counter-selections and should be used at the same concentration as AZT. Combining a targeted transformation of the tdk-kan cassette with an allelic rescue effectively allows a 2 step process for replicating any mutant allele in any competent ADP1 genomic background. To perform allelic rescue:- Follow the above protocol for knocking out your gene of interest, making sure to replace the entirety of the region you wish to exchange. Use of clones from the ADP1 knockout collection is encouraged.
- Amplify the mutant allele of your choosing from the allele containing ADP1 strain. Add 500-1000bp of flanking homology on each side of the region of interest.
- Transform the recipient strain using the amplified PCR product containing the mutant allele. Be sure to include a no DNA transformation control.
- Plate the transformation on LB + 200ug/mL AZT plates (both undiluted and a 1:100 dilution). Allow to grow overnight at 30C.
- Pick 3 colonies from the LB-AZT plates, inoculate into 5mL sterile LB and grow to confluence in a 30C shaking incubator.
- Assay for the loss of the tdk-kan cassette via PCR using the confluent culture (see above for specifics). Glycerol stock successful rescues.
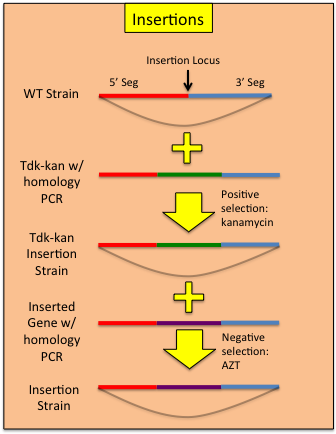
Constructing Insertions
Using a modified allelic exchange procedure, genetic constructs can be added to the ADP1 genome without the need to maintain a selective marker.- In the construction of the tdk-kan integrated strain, target the cassette to the area where you would like to perform the insertion. It is not necessary to swap out a region of the genome with the cassette.
- Rescue the tdk-kan insertion with with sequence you wish to insert.
Contributors
Brian Renda, Gabriel Suarez, Isaac Gifford Barrick Lab > ProtocolList > ProtocolsAcinetobacterOverlapPCRTransformation