Barrick Lab :: Previous Research
- Leafhoppers: Evolution and Biochemistry of Natural Nanoparticles
- Identifying Mutations that Promote Bacterial Evolvability
- Experimental Evolution with Expanded Genetic Codes
- AG3C: Associating Growth Conditions with Cellular Composition
- Discovering Functional Nucleic Acid Families by Deep Sequencing and Fold Sampling
- Evolution Experiments with Digital Organisms
Leafhoppers: Evolution and Biochemistry of Natural Nanoparticles

Leafhopper and SEM image of brochosomes on its wing
- Lay et al. (2025) ACS Synth. Biol. PMID:40052868
- Burks et al. (2023) Biomacromolecules PMID:36516996
- Li et al. (2022) Mol. Biol. Evol. PMID:36026509
Identifying Mutations that Promote Bacterial Evolvability
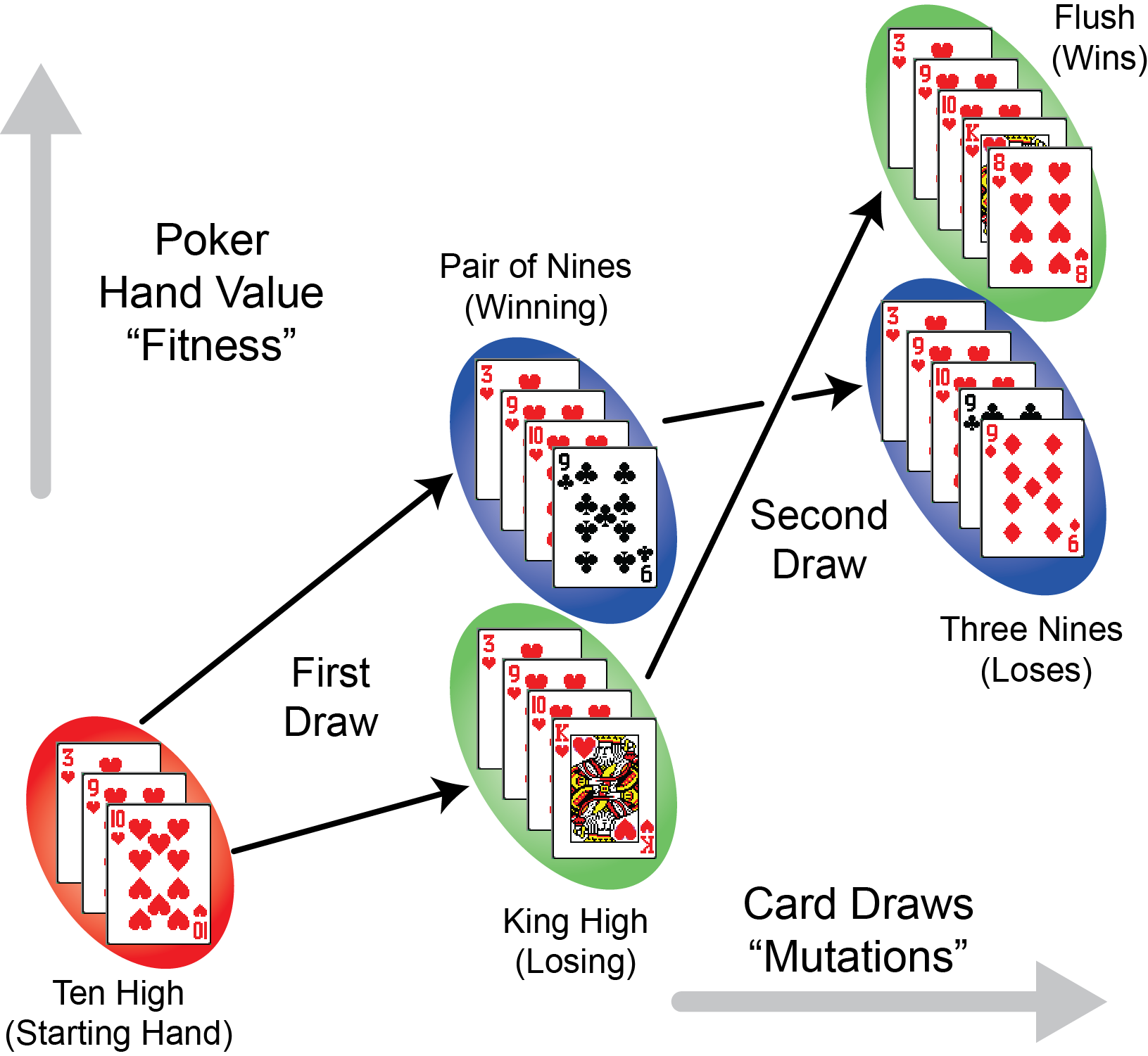
Epistasis Poker. Early draws of cards that are more beneficial to the value of a poker hand (analogous to the most beneficial mutations in the eventual loser E. coli), prevent them from reaching the best hand after more cards are drawn.
- Leon et al. (2018) PLoS Genet. PMID: 29649242
- Quandt et al. (2015) eLife PMID: 26465114
- Quandt et al. (2014) PNAS PMID:24379390
- Blount et al. (2012) Nature PMID:22992527
- Woods et al. (2011) Science PMID:21415350
Experimental Evolution with Expanded Genetic Codes
Another way to potentially increase evolvability is to augment the chemical diversity of the building blocks available to an organism. We are evolving microorganisms, including bacteriophage, in the context of expanded genetic codes that re-code the amber stop codon into various non-canonical amino acids. This change increases the sequence-structure-function space available for evolution to explore. We are studying how evolution must compensate for the global change in how the information in an organism's genome is decoded, in cases where it is possible for an organism to survive this transition, and also how dependence on the new amino acid develops over time, such that an organism can no longer survive in the context of a normal genetic code. Finally, we are systematically examining how the unique chemistries of different amino acid building blocks are tolerated and uniquely useful for adaptation and using this technology to recapitulate the steps in a hypothesized pathway in which new post-translational modifications evolve in a series of adaptive steps from nonenzymatic metabolite damage to the proteome. Representative Publication- Hammerling et al. (2014) Nat. Chem. Biol. PMID:24487692
AG3C: Associating Growth Conditions with Cellular Composition
We are part of a collaboration of eight labs (including experimental biologists, computational biologists, and mathematicians) that is collecting and analyzing comprehensive transcriptomics, proteomics, lipidomics, and metabolic flux data from bacterial samples grown under different conditions. The aim is to solve the "inverse problem" of predicting the conditions that were used to culture a microbial sample from measurements in these high-dimensional data sets (AG3C Website). Our lab is primarily responsible for creating the microbial samples and using RNA-seq to examine RNA expression levels. Representative Publication- Houser et al. (2015) PLoS Comput. Biol. PMID:26275208
Discovering Functional Nucleic Acid Families by Deep Sequencing and Fold Sampling
A final way that we are studying how to improve the potential of evolutionary methods is in systems for the in vitro selection of nucleic acid molecules. Nucleic acids with random sequences often misfold into inactive structures that are kinetically trapped. That is, they become stuck and cannot unfold to rearrange into other, potentially active, structures on a reasonable timescale. It is possible that this potential for misfolding, which is known to occur for even natural ribozymes, limits the recovery of new families of functional nucleic acids (aptamers, ribozymes, deoxyribozymes, riboswitches) by laboratory evolution methods that select for functional molecules from pools of 10131015 molecules with randomized sequences. We expect this problem to more greatly affect longer nucleic acid sequences, which have the capacity for folding into elaborate three-dimensional structures that achieve more rapid rates of catalysis or tighter binding affinities. We are currently using next-generation deep-sequencing techniques and methods to allow a single nucleic acid strand to have multiple chances to fold correctly to pass selection to see if they can enhance our ability to recover more complex families of functional nucleic acids. Representative Publications- Reba et al. (2012) Artificial Life XIII PMID:25432719
Evolution Experiments with Digital Organisms
Populations of self-replicating computer programs mutate and compete for CPU cycles in the Avida artificial life system. This environment for digital organisms has been used to ask fundamental questions that transcend the substrate that is evolving. The speed of digital evolution experiments, transparency of the underlying mechanics, and ability to manipulate and record every aspect of the evolutionary process make it possible to more readily explore cause and effect relationships in Avida than in natural systems. I have extensive experience with Avida. My lab will use Avida to complement studies in biological systems: both as a way of more clearly illustrating and exhaustively characterizing phenomena of interest and as a source of new candidate laws and insights to motivate new lines of experimental inquiry. Resources

| I | Attachment | History | Action | Size | Date | Who | Comment |
|---|---|---|---|---|---|---|---|
| |
leafhopper_website.png | r1 | manage | 478.4 K | 2025-03-23 - 15:37 | JeffreyBarrick |
{kind=link}
{kind=link}
Barrick Lab > ResearchInterests > PreviousResearch